La inmunidad es un concepto complicado que puede dividirse en dos brazos principales: los sistemas innato y adaptativo. La inmunidad innata como defensa de primera línea contra patógenos consiste en barreras físicas, factores solubles y células. La inmunidad adaptativa se compone de una gama amplia de células especiales llamadas linfocitos B y T.
La ruptura de la autotolerancia como sello de autoinmunidad se basa en la inmunidad adaptativa, pero la inmunidad innata también tiene características únicas, que la convierten en un conductor central de algunas respuestas inmunes críticas.
La autoinmunidad es una consecuencia del fracaso de autotolerancia y una reacción inmune contra un autoantígeno, que se clasifica como sistémico u órgano específico. Las enfermedades autoinmunes sistémicas son una gama amplia de trastornos que incluyen artritis reumatoide (AR), lupus eritematoso sistémico (LES), síndrome de Sjögren, polimiositis (PM) y esclerosis sistémica. Este grupo heterogéneo de trastornos se caracteriza por la presencia de autoantígenos expresados de forma ubicua y la participación de múltiples tejidos y órganos. Hasta donde se sabe, aunque la asociación entre la inmunidad adaptativa y la enfermedad autoinmune se estudió de manera extensa, la importancia de la inmunidad innata en el desarrollo de trastornos autoinmunes aún no se determina. Si se considera la falta de cura para este tipo de enfermedades, lo cual lleva a la necesidad de un tratamiento duradero, los estudios sobre la investigación básica brindan nuevos conocimientos para ayudar a identificar nuevos objetivos terapéuticos. Este estudio destaca cómo la inmunidad innata podría afectar las enfermedades autoinmunes sistémicas y presenta terapias dirigidas a componentes de la inmunidad innata en enfermedades autoinmunes sistémicas que se encuentran bajo investigación en la actualidad.Artritis reumatoide
La artritis reumatoide es una enfermedad autoinmune sistémica que afecta de forma principal a las articulaciones, lo que lleva a una inflamación crónica de articulaciones, daño del cartílago, erosión ósea y por último complicaciones sistémicas. Aunque la etiología de la AR no se delinea de forma completa, tanto el sistema inmunológico innato como adaptativo son indispensables en la patogenia de la AR. Esta revisión, se centra en células inmunes innatas y su papel crucial en la AR.
Los macrófagos (MQ) son células inmunes importantes y actores centrales en la patogenia de la AR. Ellos son la fuente principal de citocinas proinflamatorias como el factor de necrosis tumoral (TNFα), interleucina-1 beta (IL-1β) e IL-6, que generan respuestas inflamatorias y contribuyen a la destrucción de cartílago y resorción ósea en pacientes con AR. Los MQ sinoviales desempeñan un papel fundamental en los eventos que provocan inflamación, como el reclutamiento de las células inmunitarias, expansión de células de fibroblastos y secreción de proteasas, que conduce a la destrucción de la membrana sinovial. Los investigadores indican que el desequilibrio entre los MQ M1 y M2 tiene un papel fundamental en la patogenia de la AR. Los MQ en el líquido sinovial de los pacientes con AR producen grandes cantidades de TNFα e IL-1β, importantes citocinas proinflamatorias que se secretan de manera característica por los MQ M1.
Los neutrófilos son las primeras células inmunitarias que llegan al sitio de inflamación. Su función incluye la fagocitosis, la producción de especies reactivas de oxígeno (ERO) y la generación de trampas extracelulares de neutrófilos (TEN) en la defensa del huésped. Las TEN pueden ser la fuente clave de autoantígenos citrulinados, que pueden desencadenar el progreso de la AR. Los autoantígenos citrulinados en las TEN pueden ser captados por sinoviocitos similares a fibroblastos (SSF), presentados a las células T, lo que conduce a la expansión de la respuesta de células T y B en pacientes con AR. Las citocinas proinflamatorias como TNFα, IL-6, IL-8 e IL-17A pueden aumentar las TEN en los neutrófilos de la AR. Con el tiempo, las TEN estimulan una mayor producción de citocinas e inflamación mediante la activación de SSF y MQ. Los neutrófilos en el líquido sinovial de los pacientes con AR producen un estimulador de linfocitos B (BLyS) que contribuye a la activación de los linfocitos B autorreactivos; además, los neutrófilos de la articulación sinovial producen un activador del receptor del ligando del factor nuclear kappa-B (RANKL) que se implica en la activación y la diferenciación de los osteoclastos y la erosión ósea en pacientes con AR. Los inmunocomplejos son los mayores activadores de los neutrófilos en las articulaciones con AR, mediante la interacción con el receptor FCγ en los neutrófilos.
Las células dendríticas (CD), como células profesionales presentadoras de antígenos, son de forma probable, actores clave en la iniciación de inflamación articular e implicadas en el desarrollo de la AR. Hay un número aumentado de CD mieloides y plasmacitoides (CDp) en las articulaciones de pacientes con AR. Algunos estudios sugieren que en el ambiente proinflamatorio, como el líquido sinovial de pacientes con AR, la función de las CD es diferente. Estas células se propusieron como un subconjunto de CD inflamatorias. Las CD inflamatorias son el principal inductor de las células T colaboradoras (Th17) productoras de IL-17 mediante la producción de IL-23 en la sinovia de la AR. Fumar tiene algunos efectos sobre las CD, como la modificación de los antígenos que presentan las CD y la regulación de la actividad de las CD.
Las células asesinas naturales (NK) desempeñan un papel importante en la patogenia de la AR mediante la producción de citocinas inflamatorias y la interacción con varias células inmunes en el tejido sinovial. Algunos estudios indicaron el aumento de células NK en la membrana sinovial de los pacientes con AR que expresan niveles elevados de marcadores de activación y citocinas como TNFα e interferón-γ (IFNγ). Las células NK secretan IFNγ, que puede participar en la inflamación mediante la inducción de la activación de células B, cambio de clase, y maduración de las CD.
Los SSF, células no inmunes en la membrana sinovial, tienen una función crítica en la patogenia de la AR. En un microambiente inflamatorio, los SSF producen quimiocinas como CCL2, CCL5, CCL8, CXCL5 y CXCL10, las cuales reclutan monocitos y MQ, y de manera posterior contribuyen a la patogenia y la inflamación de la AR. Los SSF activados producen niveles elevados de RANKL, que es un factor significativo para la diferenciación de osteoclastos y reabsorción ósea. Los SSF funcionan como una célula presentadora de antígenos e interactúan con células T CD4+; además, los SSF tienen un papel fundamental en la diferenciación de células T con la producción de citocinas. Los SSF producen grandes cantidades de factor activador de células B (BAFF) e IL-6 que contribuyen a la maduración y la supervivencia de las células B.
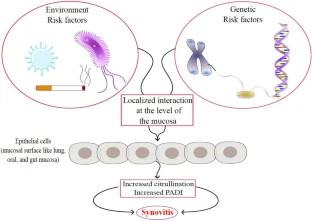
Se demostró la función significativa de las superficies mucosas en la patogenia de AR. Existe alguna evidencia de que el primer golpe en romper la autotolerancia a la AR puede originarse en las superficies epiteliales. Muchos factores de riesgo como fumar y la enfermedad periodontal, a nivel pulmonar y oral, implican autoinmunidad al impulsar la producción de autoantígenos citrulinados, y de forma consecuente anticuerpos antiproteína citrulinada (AAPC). Se confirmó una fuerte asociación entre los AAPC y el progreso de la AR en varias fases de la enfermedad en pacientes con AR. La disbiosis intestinal contribuye al estado inflamatorio mediante la polarización inducida Th17 y el desequilibrio entre Th17 y las células T reguladoras.
Muchos estudios muestran el papel patológico de las células cebadas (CC) en la AR. El número de CC se incrementa en la sinovial de la AR. Las CC activadas producen varios mediadores, citocinas y quimiocinas que reclutan células inflamatorias en la membrana sinovial. La principal fuente de IL-17A en la sinovial de la AR son las CC. Se indica que las CC mejoran la supervivencia, activación, proliferación y diferenciación de células B vírgenes. Las células T γδ se distribuyen en la mucosa y tejido epitelial de forma principal. Estas células tienen un papel importante en enfermedades autoinmunes como la AR. Las células T γδ contribuyen a producción incrementada de citocinas proinflamatorias, autoanticuerpo patogénicos, y al final conducen al inicio de esta enfermedad autoinmune.
Varios autoanticuerpos presentes en pacientes con AR, tales como anticuerpos anticolágeno tipo II, AAPC y factor reumatoide que se dirige a los antígenos del cartílago y la membrana sinovial, conducen a la formación de complejos inmunes. Estos complejos inmunes pueden activar el complemento y de forma consecuente causar la destrucción crónica de la articulación. Numerosos estudios demuestran la presencia de componentes del complemento activados o escindidos en la articulación, y elementos como los complejos C1q-C4 en la circulación de pacientes con AR. El nivel de C5a se encuentra elevado en la membrana sinovial de pacientes con AR, que se relaciona con un mayor número de neutrófilos infiltrantes. Parece que C3a y C5a se involucran en la activación de la vía del inflamasoma NLRP3, el cual es importante en el proceso inflamatorio de la AR (Figuras 1, 2).
Algunos de los objetivos relacionados con la inmunidad innata para el tratamiento de la AR se enumeran en la Tabla 1 (ensayos clínicos y aprobados por la Administración de Medicamentos y Alimentos de los EE. UU. [FDA]).
Lupus eritematoso sistémico
Lupus eritematoso sistémico (LES), también conocido de manera simple como lupus, es una enfermedad autoinmune crónica multifacética con manifestación proteica que afecta a múltiples órganos, como riñones, piel, corazón y pulmones. Se caracteriza por la presencia de autoanticuerpos específicos para autoantígenos, como ADN bicatenario (dsDNA), ribonucleoproteínas, histonas, y ciertos componentes citoplasmáticos. Algunos estudios aclararon que varias células inmunes y citocinas proinflamatorias desempeñan un papel importante en la patogenia del LES. Se indica que el sistema inmunológico innato, en particular los MQ, son actores claves en la patogénesis del LES. En esta revisión, se centra en los componentes celulares y moleculares de la inmunidad innata en la patogénesis del LES.
Los MQ son defectuosos en la fagocitosis y el aclaramiento de células apoptóticas; por lo tanto, la exposición prolongada de autoantígenos a las células inmunitarias adaptativas proporciona señales de supervivencia para células B autorreactivas y, en consecuencia, pérdida de tolerancia a antígenos nucleares liberados de células apoptóticas. La plasticidad es una característica importante de los MQ, que depende del medio de citocinas. Los MQ se clasifican en dos grupos principales: MQ activados de forma clásica (M1) y MQ activados de forma alternativa (M2). Los MQ M1 se inducen por el IFNγ y los lipopolisacáridos (LPS) que participan en las respuestas inflamatorias, mientras que los MQ M2 se inducen por la IL-4 e IL-13 que participan en la remodelación tisular. Algunos datos sugieren que los MQ M1 y M2 tienen diferentes papeles en el desarrollo del LES. Los MQ M1 aumentan la gravedad de la afección, mientras que los MQ M2 la reducen. Por tanto, la polarización de los MQ modula el desarrollo de LES. Se reportó que las citocinas polarizadoras de M2 como la IL-4 pueden tener efectos terapéuticos para reducir los síntomas del LES.
De los glóbulos blancos circulantes, los neutrófilos son los más grandes en número. Se propone que desempeñan un papel patogénico en el LES. Los pacientes con LES tienen un número elevado de neutrófilos apoptóticos en la circulación; este escenario se relaciona con el desarrollo de autoanticuerpos contra el ADN y la actividad de la enfermedad. La eliminación de las TEN es defectuosa en pacientes con LES y conduce a la presentación de autoantígenos, como el ADN inmunogénico, histonas y proteínas de neutrófilos al sistema inmunológico y contribuye al desarrollo de autoanticuerpos y citocinas proinflamatorias, que impulsan la patogenia del LES. Los neutrófilos de los pacientes con LES tienen una función anormal; la capacidad de su fagocitosis se reduce, mientras que su producción de ERO se aumenta. La TENosis, un tipo de muerte celular, se eleva en los neutrófilos de pacientes con LES por la presencia de complejos anticuerpo-ribonucleoproteína, que activan varias células inmunes. Los modelos de ratón de LES demuestran TENosis y autoanticuerpos elevados de la médula ósea que distinguen a los componentes de las TEN. Se propuso que se requiere endonucleasa DNasa1 para degradar las TEN. Algunos pacientes con LES tienen inhibidores de DNasa1, mientras que otros pacientes con LES tienen niveles altos de anticuerpos que se unen a las TEN y las protegen de la DNasa1. Un subconjunto distinto de células similares a los neutrófilos denominado granulocitos de densidad baja (GDB) se distingue en pacientes con LES, los cuales producen exceso de citocinas proinflamatorios como IL-6, IL-8, TNFα e IFN-I. Se propone que los GDB tienen un papel importante en la patogenia del LES.
Algunos estudios documentan que las CDs desreguladas desempeñan un papel fundamental en el inicio y desarrollo del LES. Las CD de pacientes con LES muestran una considerable reducción de la expresión de PD-L1 durante la enfermedad activa, mientras que la expresión de CD80/CD86 es elevada.
Las CD plasmocitoides (CDp) en pacientes con LES producen niveles elevados de IFNα que provoca un bucle de retroalimentación positiva en la activación de la inmunidad innata y adaptativa. Los números de las CDp disminuyen en la sangre de pacientes con LES, pero las CDp se acumulan en la piel dañada de pacientes con lupus. Varios informes señalan que el agotamiento de las CDp reduce la activación y la expansión de las células inmunes, limita la producción de autoanticuerpos, y restringe la inflamación renal en pacientes con LES.
Los niveles elevados de IFNα en pacientes con LES se correlacionan con la actividad y la gravedad de la enfermedad. La producción sostenida de IFNα, una firma del lupus, puede conducir al desarrollo de células T y B autorreactivas.
Las células NK pueden tener un papel crucial en la patogenia de LES. Se informa que el número de células NK en pacientes con LES está disminuido y su citotoxicidad deteriorada. Algunos estudios sugieren una disminución en el número de células T asesinas naturales (NKT) en pacientes con LES. La IL-15 es una citocina que desempeña un papel importante en la diferenciación y supervivencia de las NK y se eleva en pacientes con LES, escenario que se relaciona a la actividad de la enfermedad. El receptor activador CD69 se sobreexpresa en células NK de pacientes con LES con enfermedad activa. Algunos estudios indican que las células NK producen niveles elevados de IFNγ en pacientes con LES con enfermedad activa y esto se asocia con citotoxicidad y contribuye a la desregulación del vínculo entre inmunidad innata y adaptativa en el LES.
Los basófilos se involucran en las lesiones cutáneas en pacientes con LES y tienen un papel en la promoción del daño tisular. Los basófilos se proponen como biomarcador de la actividad de la enfermedad en el LES. Estas células tienen un papel importante en la inflamación y la producción de anticuerpos antinucleares por las células B. La prostaglandina D2 (PGD2) aumenta en pacientes con LES e interactúa con los receptores PGD2 en la superficie de los basófilos, lo que lleva a la migración de los basófilos a los órganos linfoides secundarios.
El sistema del complemento desempeña un papel controvertido en la patogenia del LES. Este sistema tiene funciones protectoras y patológicas. El complemento ejerce su papel de protección mediante la eliminación de complejos inmunes y células apoptóticas, así como inducción de tolerancia. La activación del complemento que conduce a la respuesta inflamatoria y daño tisular define el papel patológico de complemento. La deficiencia del complemento se correlaciona con la patogenia del LES. Deficiencias de C1q, como los defectos genéticos o autoanticuerpos anti-C1q, pueden causar LES en 90% de los pacientes. Se encuentra bien establecido que C1q se involucra en la regulación de la diferenciación de las células inmunes y la polarización de los MQ a un fenotipo tolerogénico.
Los queratinocitos pueden ser un actor clave en la patogénesis de LES. Estas células se activan con luz ultravioleta y producen citocinas inflamatorias que resultan en el reclutamiento de células inmunes y el inicio de respuestas inflamatorias (Figura 3).
Algunos de los objetivos del tratamiento relacionados con la inmunidad innata del LES se enumeran en la Tabla 2 (ensayos clínicos).
Síndrome de Sjögren
El síndrome de Sjögren (SS) es una enfermedad autoinmune sistémica que afecta a las glándulas exocrinas, afecta de forma principal las lagrimales y salivares, lo que provoca sequedad de ojos y boca. El SS se clasifica en dos formas: primaria (SSp) y secundaria (SSs). El SS primario a menudo se asocia con disfunción en el flujo lagrimal y salival y presenta un rango amplio de manifestaciones sistémicas y específicas de órganos, mientras que la SSs ocurre en asociación con otra enfermedad autoinmune, como LES o AR. Si bien la patogenia del SSp no se comprende bien en la actualidad como otras enfermedades autoinmunes, los sistemas inmunitarios tanto innato como adaptativo desempeñan un papel fundamental en la patogénesis de la enfermedad. Muchos tipos de células innatas se implican en el SS.
Algunos estudios también demuestran que los MQ son mediadores fundamentales de la patogenia del SS. La presencia de MQ M1 y M2 se muestra en las glándulas salivales en modelos múridos de SS. La polarización proinflamatoria M1 es el fenotipo principal de los MQ en el SS y las concentraciones sistémicas y locales de IL-6 aumentan de manera importante en pacientes con SS. Además, los pacientes con SS con enfermedad más activa muestran niveles más altos de IL-12, mientras que los pacientes con enfermedad menos activa muestran niveles más altos de IL-35. Los niveles de proteína 1 quimioatrayente de monocitos (CCP-1/CCL2), IFNγ, y citocinas proinflamatorias o quimiocinas que se secretan por los monocitos y los MQ, como IL-6, IL-18, IFN-I y BAFF, se elevan de forma significativa en pacientes con SS. También se reportó que disminuye el nivel de IκBα en los monocitos en el SS, lo que conduce a una desregulación de la vía de transmisión de señales del NFκB y a la producción de citocinas proinflamatorias.
Las CD desempeñan un papel importante en el SS, ya que funcionan como células presentadoras de antígenos en los centros germinales ectópicos en la glándula salival. Algunos estudios sugieren que las CD se elevan en el tejido salival de los pacientes con SSp comparado con los controles. Las CD plasmacitoides, un subconjunto específico de CD, se activan por receptores tipo Toll (TLR) y producen varias citocinas proinflamatorias, como el IFNα.
Las CD primarias son la principal fuente de IFN-I en respuesta a ácidos nucleicos extraños. IFN-I podría involucrarse en la patogenia de SS. Ácidos autonucleicos en forma de complejos de autoanticuerpos y fragmentos de células apoptóticas están presentes en pacientes con SSp y activan de manera considerable la producción de IFN-I por las CDp. Se demostró que las CDp desempeñan un papel crítico en la patogenia de SSp. El fenotipo activado y una mayor producción de citocinas proinflamatorias por las CDp-SSp puede afectar de forma dramática la inflamación de las glándulas salivales.
Algunos estudios demostraron que las células NK se involucran en la patogenia del SS, aunque el papel preciso de éstas se desconoce. Las células NK se relacionan con la inflamación de las glándulas salivales en pacientes con SSp. Las células NK expresan NCR3/NKp30 que regula la secreción y correlación de IFN-II con las CD. En pacientes con SSp, la expresión de NKp30 se eleva en comparación con los controles. Las células epiteliales de la glándula salival expresan el ligando NKp30 y la interacción de este ligando con NKp30 conduce a la producción de citocinas Th1. En contraste con la glándula salival, en pacientes con SSp, el número y la actividad de las células NK de sangre periférica se reducen de manera significativa en comparación con controles sanos. Es posible que las células NK sean protectoras en las primeras etapas de la enfermedad y luego desempeñen un papel patogénico en la enfermedad avanzada. Se comprobó que las células T asesinas naturales invariantes (iNKT) se redujeron de forma significativa en el SSp, y se sugirió una posible correlación entre el número bajo de iNKT y la lesión tisular autorreactiva.
Se sabe que, en pacientes con SS, eran normales las funciones de los neutrófilos como la fagocitosis, quimiotaxis y la quimiocinesis, mientras que se altera su capacidad de adherencia.
Algunos estudios informan un papel significativo de las células epiteliales de la glándula salival en la patogenia de SS. Además, se demostró que las células epiteliales de la glándula salival expresan niveles elevados de TLR2, TLR3 y TLR4; por lo tanto, contribuyen a la inducción de respuestas inmunes innatas al reconocimiento de patógenos extraños.
Algunas de los objetivos relacionados con la inmunidad innata para el tratamiento del síndrome de Sjögren se enumeran en la Tabla 3 (ensayos clínicos).
Polimiositis
La polimiositis (PM) es una enfermedad inflamatoria crónica inusual del tejido conectivo que involucra músculos que se atrofian con el tiempo, por lo que los pacientes con PM no pueden subir escaleras o incluso caminar. La afectación muscular es diferente en diferentes partes del cuerpo y la debilidad muscular puede conducir a problemas para los pacientes como disfagia y dificultad para respirar. La PM es una enfermedad autoinmune sin una etiología clara. Al igual que con las otras enfermedades autoinmunes, la PM es más común en mujeres. La genética se asocia con la enfermedad de manera probable, y la presencia de genes específicos del antígeno leucocitario humano (HLA) como los alelos DRB1*0301 aumenta la probabilidad de PM. La PM se asocia de manera fuerte con otras enfermedades inflamatorias, virales y cancerosas. Aunque los músculos no suelen expresar MHC I, en la PM expresan de manera extensa MHC I e incluso MHC II. Las células T citotóxicas (CTC) atacan las fibras sanas de los músculos que expresan MHC I, lo que parece ser la principal causa de lesión tisular. El aumento de la expresión de moléculas coestimuladoras ayuda a la activación de las CTC, pero no las moléculas coestimuladoras clásicas. Los músculos tienen sus propias moléculas coestimuladoras llamadas BB-1. Las biopsias musculares muestran CTC que migran a la lámina basal donde se acumulan para atacar las fibras musculares. El mecanismo por el cual las fibras se destruyen es mediante perforina y granzima, a pesar de que las fibras musculares expresan Fas. Existe poca evidencia de participación de la inmunidad innata en la inmunopatología de la PM.
Muchas citocinas inflamatorias aumentan en el suero de pacientes con PM, como IL-1, IL-2, IL-6 e IL-10 ,así como TNFα, IFN-γ y TGFβ. IL-1 y TNFα tienen un efecto devastador sobre los músculos; TNFα induce un efecto proteolítico mediante glucocorticoides, mientras que la IL-1β ejerce su efecto mediante una vía independiente de los glucocorticoides. También de observó una sobreexpresión de quimiocinas, como CXCL8, CCL9, CCL2, CXCL9 y CXCL10.
Se observaron TLR aumentados en biopsias de miopatías, en especial TLR3 y TLR7, que se activan por los ácidos nucleicos. Las células musculares necróticas pueden activar TLR3 que desencadena la producción de una gran cantidad de IL-6 de los mioblastos, lo que lleva al mantenimiento de la respuesta inflamatoria en los músculos.
La proteína del cuadro 1 del grupo de alta movilidad (HMGB1) es una proteína nuclear que puede secretarse por células inmunitarias en condiciones inflamatorias. Se secreta por los MQ y las CD en el tejido conectivo y mejora la expresión del MHC I al unirse al TLR4 en las fibras musculares.
También se estableció el papel de los eosinófilos en las miopatías; los pacientes que tienen eosinofilia en las biopsia de músculo, en especial el endomisio, tienen más fibras necróticas.
Las CD se acumulan en el tejido muscular para presentar antígenos, y las CD plasmacitoides secretan IFN-I. El IFN-I desempeña un papel esencial en la PM. Uno de los mecanismos que conducen a la secreción de IFN es mediante catelicidinas, que incluyen la LL-37, la cual es la única catelicidina expresada en los humanos. Junto con todas sus funciones antimicrobianas, antiinflamatorias e incluso funciones proinflamatorias, la LL-37 aumenta el IFN-I. Se observaron huellas de LL-37 en enfermedades autoinmunes e inflamatorias. El contacto continuo del sistema inmunológico con IFN-I puede causar insuficiencia de tolerancia inmune y enfermedades autoinmunes como la miositis.
Esclerosis sistémica
La esclerosis sistémica (ES, esclerodermia) es una enfermedad autoinmune crónica, heterogénea. El engrosamiento de la piel, la fibrosis del tejido conectivo y la disfunción de los vasos son resultados de una secreción excesiva de colágeno de los fibroblastos y su depósito, que a veces puede afectar órganos internos. Los miofibroblastos son la forma activa de los fibroblastos que segregan colágeno de forma persistente y se encuentran en las lesiones fibróticas. La principal causa de la ES aún es poco clara, pero el sistema inmunológico tiene una importante función y en la mayoría de los pacientes se encuentran autoanticuerpos. Las células T, en especial las Th2, interactúan con los fibroblastos mediante citocinas profibróticas como IL-4, IL-6 e IL-13, lo que resulta en agravamiento de la fibrosis El sistema inmunológico innato desempeña un papel fundamental tanto al inicio como en la progresión de la enfermedad.
Se demostró el papel de muchos receptores de reconocimiento de patrones (RRP) en enfermedades autoinmunes como los TLR en la ES. Los TLR identifican patrones moleculares asociados a daños (DAMPs) que se liberaron de células endógenas, por ejemplo, pueden secretarse en respuesta al estrés o daño; también la respuesta de los TLR a patrones moleculares asociados a patógenos (PAMPs) que conduce a la activación de una vía de transmisión intracelular de señales, y la respuesta inadecuada y excesiva de los TLR a sus ligandos pueden involucrarse al inicio y la exacerbación de la enfermedad. Los monocitos CD14+ y las CD se activan por TLR4, lo que resulta en una gran cantidad de secreción de CCL8 e IL-10 en pacientes con ES. CCL8 e IL-10 son factores quimioatrayentes de células T y fibróticos que aumentan en el suero de pacientes con esclerodermia. El amiloide A sérico (AAS), es un ligando para TLR2 y un tipo de familia de DAMP, puede elevarse en pacientes con esclerodermia. Esta vía conduce a la producción de IL-6 mediante el NF-кB, y la IL-6 aumenta de forma directa la secreción de colágeno de los fibroblastos. La diferenciación de fibroblastos en los miofibroblastos es común en pacientes con esclerodermia, lo que da lugar a un aumento de TLR9 ya que 60% de los miofibroblastos expresan TLR9. Cuando el TLR9 se activa por su ligando, CpG, la secreción de TGFβ aumentaría. El TGFβ tiene un papel importante en la diferenciación de los miofibroblastos y la producción de colágeno. MyD88 tiene un papel clave en la transmisión de señales TLR y patogénesis de la ES, y los investigadores podrían disminuir la fibrogénesis mediante la inhibición del MyD88.
Los monocitos son células importantes en la ES. Estas células tienen un papel importante en la remodelación de la matriz extracelular (MEC) en pacientes con ES mediante el TLR8 (ssRNA) y el inhibidor tisular de secreción de metaloproteinasa-1 (TIMP-1). Los fibroblastos degradan la MEC al secretar metaloproteinasa-1 de la matriz (MMP-1) y manteniene la homeostasis; TIMP-1 inhibe MMP-1 y conduce a la acumulación de MEC en pacientes con ES. El número de monocitos y MQ aumenta en la sangre periférica y los sitios de inflamación en pacientes con esclerodermia; esto empeora la situación debido a que estas células son resistentes a la apoptosis y a factores fibróticos liberadores. También se reportó que IL-4, IL-13 e IL-10 son más altas de lo normal en el suero de pacientes con esclerodermia, y esto provoca la formación de MQ M2, lo que significa más TGFβ y más fibrosis.
Durante años, los investigadores intentaron encontrar un vínculo entre ES y el aumento o disminución de componentes del complemento. Parece que la presencia de autoanticuerpos ayuda a activar el complemento y los factores de activación aumentan en las tres vías. El daño vascular del endotelio es una causa común de esclerodermia, que parece ser un elemento importante en la inducción de la enfermedad. El factor H es un regulador del complemento y su principal función es proteger las células huésped contra el complemento. Las células hospedadoras pueden resultar dañadas por la disfunción del factor H y liberar contenido celular, reaccionar con autoanticuerpos existentes, y formar complejos inmunes.
El inflamasoma es un conjunto de múltiples factores que se activa de forma principal por TLR que conducen a la caspasa-1 activada, lo que resulta en la activación de IL-1β e IL-18 por caspasa-1. El NLRP3 es el inflamasoma más conocido y tiene el papel principal en muchas enfermedades autoinflamatorias y autoinmunes. El papel de IL-1β e IL-1α se confirmó en la fibrosis por los efectos autocrinos sobre los fibroblastos, y los niveles séricos de IL-1β son más altos en pacientes con ES que en controles sanos. Contrario a todos los esfuerzos, el papel de IL-18 aún no se aclara en la patogénesis de la ES. La IL-18 parece tener un efecto antifibrótico a diferencia de la IL-1β pero aumenta en pacientes con ES.
La acumulación de células cebadas se observó en la piel afectada de pacientes con ES y tiene un papel importante en las enfermedades asociadas con fibrosis porque las células cebadas tienen gránulos densos que contienen citocinas proinflamatorias, TGFβ e histamina que ayudan a formar miofibroblastos. Las células cebadas y los miofibroblastos interactúan mediante uniones comunicantes o envían vesículas y se ayudan entre sí para aumentar la inflamación.
Algunos de los objetivos del tratamiento relacionados con la inmunidad innata de la ES se enumeran en la Tabla 4 (ensayos clínicos).
Conclusión
Existe una gran necesidad insatisfecha de identificar cómo inicia, progresa y se propaga la autoinmunidad. En este contexto, la inmunidad innata actúa tanto como proveedora de condiciones inflamatorias para la función de la inmunidad adaptativa así como parte independiente o copartícipe del sistema inmunológico adaptativo. La desregulación de la inmunidad innata se demuestra en muchas enfermedades, por lo que la manipulación de las vías relacionadas hasta ahora llama la atención, en la medida que la focalización del TNFα revolucionó el manejo y los resultados de la enfermedad de AR. Además, en la actualidad se encuentran en prueba una gran cantidad de otras terapias en ensayos clínicos; sin embargo, aún queda mucho por aprender sobre este tema y los conceptos erróneos relacionados.





No hay comentarios:
Publicar un comentario
Nota: solo los miembros de este blog pueden publicar comentarios.