El término vasculitis se refiere a un grupo de afecciones cuya fisiopatología se media por la inflamación de los vasos sanguíneos. La mayoría de las vasculitis son sistémicas y pueden presentar manifestaciones clínicas variables, lo que requiere un abordaje multidisciplinario. Se reportan varias etiologías; de forma independiente a la afectación del órgano específico, la vasculitis puede ser primaria o secundaria a otra enfermedad autoinmune o puede asociarse con otros precipitantes como fármacos, infecciones o malignidad.
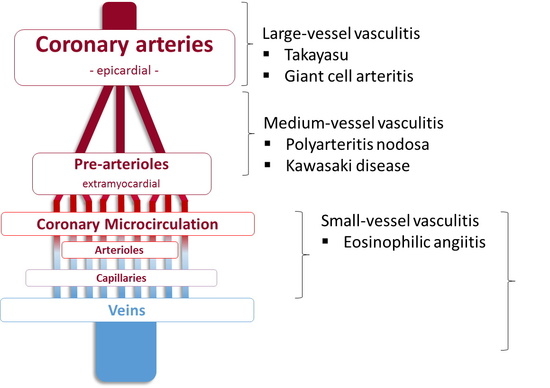
2. Entidades de vasculitis más frecuentes con afectación coronaria
Aunque una discusión exhaustiva sobre las características clínicas, la fisiopatología y el tratamiento de la vasculitis va más allá del alcance de este artículo y se centra en la afectación coronaria, los siguientes párrafos describirán en forma breve las características principales de cada condición asociada con la afectación coronaria, que también se resumen en la tabla 1.
2.1. Vasculitis de vasos grandes
2.1.1. Takayasu
La arteritis de Takayasu incide con mayor frecuencia a mujeres jóvenes y afecta a grandes vasos, por lo general el arco aórtico y las ramas proximales de la aorta, así como las arterias pulmonares. El curso de la enfermedad suele ser crónico, con síntomas cutáneos, neurológicos, gastrointestinales y constitucionales. La angina de pecho puede ocurrir después de la estenosis ostial de la arteria coronaria por aortitis o arteritis coronaria en 10-45% de los casos y puede tener secuelas clínicas graves, aunque es posible la regresión tras la inmunosupresión. En estos casos, la estenosis de la arteria coronaria es causada por la difusión del proceso inflamatorio y la proliferación de la íntima de la pared de la aorta ascendente. La patogenia no está clara, pero se cree que se involucran los mecanismos mediados por células (de forma particular los linfocitos citotóxicos, en especial los linfocitos T gamma delta que liberan la proteína citolítica perforina). La presencia de un antígeno específico (aún desconocido) en la aorta podría explicar la localización selectiva de la enfermedad. En la histología, están presentes células mononucleares, de forma predominante linfocitos, histiocitos, macrófagos y células plasmáticas, y se encuentran células gigantes e inflamación granulomatosa en la media arterial. La expansión de estos procesos inflamatorios puede llevar por un lado a la destrucción de la lámina elástica, lo que provoca la dilatación aneurismática del vaso afectado, o al estrechamiento de la luz. Los marcadores inflamatorios sistémicos suelen elevarse. La terapia sistémica consiste en corticoesteroides e inmunosupresión, pero la terapia anti-IL6 con tocilizumab también se asocia con una reducción de los marcadores de inflamación y, lo que es más importante, también del número de lesiones de las arterias coronarias. Takayasu es una enfermedad crónica progresiva con remisiones y recaídas y una supervivencia de 80 a 90% a los cinco años.
2.1.2. Arteritis de células gigantes
Esta condición comparte varias similitudes con la arteritis de Takayasu, y el diagnóstico diferencial se basa en un criterio más bien artificial de edad (mayor de 50 años en la arteritis de células gigantes y menor de 40 en Takayasu) y afectación del distrito vascular (afectación más frecuente de los vasos supraaórticos en la arteritis gigante). Tanto la arteritis de células gigantes como la de Takayasu afectan la aorta y sus ramas principales y son indistinguibles en histología. Aunque se cree que la prevalencia de afectación de las arterias coronarias en la arteritis de células gigantes es menor, estudios de cohortes grandes reportan que la incidencia de infartos agudos de miocardio es dos veces mayor que la de los pacientes sin arteritis de células gigantes, en particular al comienzo del diagnóstico (cociente de riesgo de IAM: 11.9 [2.4-59.0]). Se recomienda el tratamiento con tocilizumab, un anticuerpo anti-IL6, además de los corticoesteroides, pero no se investigó su eficacia de forma específica en la vasculitis coronaria. La enfermedad no compromete la esperanza de vida, excepto en aquellos pacientes en los que hay disección aórtica.
2.2. Vasculitis de vasos medianos
2.2.1. Poliarteritis nodosa
La poliarteritis nodosa es una vasculitis necrotizante sistémica que afecta de forma predominante a arterias de tamaño mediano, de manera más rara a arterias musculares pequeñas en adultos de mediana edad. A diferencia de la poliarteritis microscópica, la granulomatosis con poliangeítis, la poliarteritis nodosa tiene anticuerpos anticitoplasma de neutrófilos (ANCA) negativos. Puede afectar de forma virtual a cualquier órgano del cuerpo (riñones, piel, articulaciones, músculos, nervios y tracto gastrointestinal), en general en combinación, pero de forma usual no afecta los pulmones. La afectación coronaria se considera rara, pero puede ser grave, incluida una enfermedad agresiva de tres vasos con un resultado prematuro (figura1). Las secuelas clínicas de esta afectación incluyen insuficiencia cardíaca congestiva, hipertensión, pericarditis y arritmias.
La afectación coronaria puede manifestarse en forma de estenosis, oclusión, aneurisma o disección. En una revisión reciente de casos, se identificaron un total de 34 pacientes con una edad promedio de 41 años a partir de 32 publicaciones. El sexo masculino es más frecuente, la enfermedad coronaria fue la primera manifestación de poliarteritis nodosa en 3/4. El curso clínico de la enfermedad fue en general muy grave, con casos de muerte por parada cardiaca, edema pulmonar con hemorragia alveolar o hemorragias intracraneales múltiples tras el tratamiento trombolítico. Se cree que la formación de complejos inmunes como resultado de una infección por virus (virus de la hepatitis B o C) y la leucemia de células pilosas median la reacción inflamatoria, que con mayor frecuencia conduce a un engrosamiento y estenosis de la media más que a la formación de un aneurisma. La terapia médica incluye ciclofosfamida además de corticoesteroides y/o azatioprina, aunque la efectividad a largo plazo de este enfoque parece ser limitada. La supervivencia a cinco años alcanza 80%.
2.2.2. Kawasaki
También conocida como síndrome de los ganglios linfáticos mucocutáneos, la enfermedad de Kawasaki es una vasculitis sistémica aguda, autolimitada de vasos pequeños y medianos con una afectación frecuente de las arterias coronarias (la derecha más a menudo que la izquierda) que afecta a niños menores de 5 años con casos raros en niños mayores. Aunque la enfermedad de Kawasaki en general tiene un curso febril autolimitado, dada la incidencia decreciente de enfermedad reumática, representa la causa más común de cardiopatía adquirida en la infancia en los países desarrollados y representa hasta 5% de los casos de síndrome coronario agudo en pacientes menores de 40 años. De manera clínica, se presenta como erupción polimorfa, cambios en las mucosas (que incluyen labios secos y agrietados y lengua de fresa), cambios en las extremidades (que incluyen eritema palmar y/o plantar, hinchazón y descamación), conjuntivitis no purulenta bilateral y linfadenopatía cervical (≥1.5 cm de diámetro), por lo general unilateral. Aunque también se plantea la hipótesis de una influencia genética, como en el caso de la enfermedad de Behçet, la patogénesis de Kawasaki no está clara y puede relacionarse con un patógeno transmitido por el viento o el agua. El grupo de edad más común afectado son los niños menores de cinco años, pero los casos en adultos también son comunes. Por ejemplo, de manera reciente se reportó un caso de síndrome de choque de la enfermedad de Kawasaki de inicio en la edad adulta complicado por aneurismas coronarios en un hombre de 20 años de ascendencia asiática oriental. Como en el caso de la enfermedad de Behçet, la prevalencia es mayor y el pronóstico peor en los varones afectados por Kawasaki. En la hipótesis actual, se cree que la activación de una reacción inmune que involucra en la primera fase a los neutrófilos y luego a los linfocitos, citocinas y proteinasas; el factor de necrosis tumoral alfa (TNF-a); las interleucinas 1, 4 y 6 y las metaloproteinasas de la matriz (MMP3 y MMP9) desencadenadas por la exposición a un virus transportado por el aire pueden sustentar la enfermedad de Kawasaki. Las células T CD8+, las células plasmáticas y los monocitos provocan la liberación de citocinas proinflamatorias IL-1β y TNFα. Estos procesos pueden evolucionar durante meses o años, lo que resulta en una arteritis crónica. Las células plasmáticas IgA oligoclonales parecen ser centrales en la cascada que conduce a la arteritis coronaria. Las manifestaciones clínicas pueden incluir miocarditis y arteritis que provocan necrosis fibrinoide de la lámina elástica interna y la formación subsiguiente de aneurismas coronarios en hasta un tercio de los pacientes no tratados. Los aneurismas cerebrales son menos frecuentes en 1 a 2% de los pacientes. Los monocitos, neutrófilos y macrófagos parecen implicarse en la patogenia de estas lesiones vasculares. Como resultado de estos procesos inflamatorios, una respuesta de curación inapropiada también puede causar estenosis coronaria. De lo contrario, las complicaciones típicas asociadas con la presencia de los aneurismas incluyen la formación de trombos que provocan embolia y oclusión y rotura periférica. También se reportan casos de regresión de pequeños aneurismas coronarios tras la terapia inflamatoria. En la fase aguda de la enfermedad de Kawasaki, se recomienda la terapia con inmunoglobulina intravenosa, corticoesteroides y monoterapia con aspirina. La terapia con inmunoglobulinas y corticoesteroides se asocia con una reducción en la incidencia de eventos coronarios (razón de momios: 0.3 [0.2-0.5]), pero la presencia de signos compatibles con vasculitis coronaria es un predictor negativo de la capacidad de respuesta a las inmunoglobulinas. El pronóstico es variable y depende del tamaño de los aneurismas. Por tanto, es importante realizar un seguimiento regular.
2.3. Vasculitis de vasos pequeños
Angitis eosinofílica
Las vasculitis asociadas a anticuerpos anticitoplasma de neutrófilos (ANCA) componen una familia de afecciones que involucran vasculitis sistémica grave de vasos pequeños con autoanticuerpos dirigidos contra las proteínas de los neutrófilos leucocitos proteinasa 3 o mieloperoxidasa. Su presentación clínica y terapias se revisaron de manera reciente. Dentro de este grupo de enfermedades, la granulomatosis eosinofílica con poliangeítis afecta con mayor frecuencia a los vasos coronarios. Esta condición (que no está mediada en forma necesaria por ANCA) tiene una incidencia de ~ 0.5 a 2 casos por millón por año, su momento de aparición es típica en la edad media o avanzada y es igual de frecuente en hombres y mujeres. De forma histológica, se caracteriza por una inflamación granulomatosa necrosante y rica en eosinófilos que afecta de manera predominante a vasos pequeños a medianos; la eosinofilia es el marcador típico y la afectación pulmonar y el asma a menudo se asocian. Conectado, pero no superpuesto, con esta condición se notificó el primer caso de una nueva arteritis de tamaño mediano con inflamación eosinofílica limitada a la adventicia y el tejido blando periadventicial de las arterias coronarias epicárdicas en 1989. La periarteritis coronaria eosinofílica no es sistémica y se diferencia de la poliarteritis nudosa y el síndrome de Churg-Strauss en que no afecta a otras capas arteriales y no muestra necrosis fibrinoide. Esta forma de vasculitis coronaria tiene un inicio en la edad adulta y se asocia con casos de disección coronaria espontánea o espasmo coronario. La arteritis eosinofílica sin disección es más frecuente en hombres que en mujeres, mientras que una disección espontánea, predominante de la coronaria descendente anterior izquierda, es más frecuente en mujeres. Aunque la etiología y patogenia de esta afección son inciertas, es posible que esta enfermedad pueda desempeñar un papel en varios casos de disección espontánea que permanecen sin diagnosticar de manera etiológica. El síndrome de Churg-Strauss es una enfermedad relacionada que se diagnostica en presencia de los siguientes seis criterios: asma grave, infiltrados pulmonares fugaces en las radiografías de tórax, eosinofilia, anomalías del seno paranasal, infiltración eosinofílica en la biopsia y manifestaciones neurológicas. La mayor incidencia se da en la cuarta-quinta década de la vida y es mayor en las mujeres. La afectación cardíaca puede manifestarse como pericarditis, miocardiopatía restrictiva o dilatada, miocarditis, arritmias y muerte súbita cardíaca. El pronóstico de estas enfermedades mejora de forma notable con el uso de corticoesteroides, y la mortalidad está en el rango de 30% a los 5 años.
2.4 Otras formas
2.4.1. Behçet
La enfermedad de Behçet es una enfermedad grave más frecuente en el Mediterráneo oriental y el este, donde alcanza una incidencia de 0.03% de la población. La incidencia de masculinos, de manera más común en la cuarta y quinta década de la vida, es tres veces más alta que en mujeres. Las manifestaciones más comunes incluyen signos de activación inflamatoria como fiebre y síntomas constitucionales. Las características clínicas de esta enfermedad incluyen ulceraciones orales y/o genitales, enfermedad ocular, y artritis. La enfermedad de Behçet tiene una causa desconocida a pesar de que se sospecha una predisposición genética; su fisiopatología se basa en mecanismos autoinflamatorios, daño/disfunción endotelial, y fibrinólisis defectuosa que involucra la activación de linfocitos T, como las células T cooperadoras 17, formación de complejos inmunes, activación de neutrófilos, y la secreción de citocinas inflamatorias. Desde la perspectiva inmunológica, Behçet se asocia con perivasculitis con infiltración neutrofílica, necrosis fibrinoide e hinchazón endotelial. Los anticuerpos antilinfocitos y anticardiolipina son una característica de esta condición y permite su diagnóstico. El involucro arterial en la enfermedad de Behçet se manifiesta por sí mismo como aneurisma de las arterias de talla mediana y grande. Las manifestaciones vasculares de Behçet varían entre 10 y 30% de los casos y quizá involucren ambas arterias y, de manera aproximada 4 veces más frecuente, venas. El involucro vascular presagia un pronóstico negativo, en particular en hombres más jóvenes sin factores de riesgo tradicional de enfermedad de las arterias coronarias (excepto por el tabaquismo). Aunque la enfermedad coronaria nativas se describe de manera más común, se describen casos de reestenosis en el stent. La terapia puede incluir colchicina y terapia inmunosupresora general con corticoesteroides, azatioprina, ciclosporina A, y ciclofosfamida. El pronóstico es bueno, aunque la incidencia de recaídas es muy elevada.
2.4.2. Enfermedad de Erdheim-Chester
La enfermedad de Erdheim-Chester es una histiocitosis poco común que no es de Langerhans y es de forma esencial una neoplasia maligna de las células progenitoras mieloides causada por una mutación somática de genes de moléculas de transmisión de señales, lo que explica el aumento de la expresión de citocinas inflamatorias (IL-6, interferón alfa, MCP-1). Se reportan menos de 1000 casos de esta enfermedad, más común bajo la forma de lesiones escleróticas multifocales de los huesos. El dolor óseo, los síntomas neurológicos y los síntomas cardíacos son las manifestaciones más frecuentes. Puede resultar en una afectación multisistémica que incluye fenómenos similares a vasculitis de arterias grandes y medianas, en particular de la aorta (alrededor de 60% en una serie de casos recientes). La afectación de la arteria coronaria es bastante común (55%), lo que provoca alrededor de un tercio de las muertes. La presentación más común incluye infiltración, en particular a nivel de la aurícula derecha; También son frecuentes la fibrosis periarterial, el engrosamiento y el derrame pleural. Su sello histológico es la fibrosis perirrenal. El tratamiento puede incluir un inhibidor de BRAF (vemurafenib), un inhibidor de MEK, interferón alfa o inmunosupresión con corticoesteroides o terapias citotóxicas. La supervivencia a cinco años alcanza 70%.
2.4.3. Enfermedad relacionada con IgG4
La enfermedad relacionada con la inmunoglobulina G4 es una enfermedad fibroinflamatoria rara con infiltración linfoplasmocítica multiorgánica que afecta con mayor frecuencia a varones. Todos los órganos pueden afectarse y la afectación extracardíaca puede manifestarse como sialoadenitis, tiroiditis, nefritis, linfadenopatía y enfermedad pulmonar. La patogenia de esta enfermedad parece depender de una respuesta inmune mediada por células Th1 y Th2, mientras que el papel de los anticuerpos IgG4 sigue sin estar claro. Las manifestaciones cardiovasculares de la ER-IgG4 pueden incluir aortitis, arteritis de vasos de tamaño mediano, enfermedad vascular pulmonar, flebitis, valvulopatía, pericarditis y vasculitis que se asocia a anticuerpos citoplasmáticos antineutrófilos y con mayor frecuencia involucran la vasculatura abdominal, aunque también se reportan casos cardíacos. La terapia con glucocorticoides conduce a una remisión en 98% de los casos, pero a menudo también se observa una remisión espontánea sin terapia. Sin embargo, también existen casos que son refractarios a los esteroides y requieren inmunosupresión.
3. Epidemiología
La relativa rareza de estas afecciones, así como la falta de estudios de imágenes prospectivos estandarizados que evalúen de forma sistemática la vasculatura coronaria, complican el diagnóstico de vasculitis de las arterias coronarias en pacientes con enfermedades autoinmunes conocidas. La afectación de la microvasculatura coronaria (no visible en la tomografía computarizada o angiografía) y el hecho de que la vasculitis de la arteria coronaria puede manifestarse en forma de (micro)espasmo vascular coronario en lugar de estenosis fija complican aún más el diagnóstico y pueden dar lugar a muchos falsos negativos. Al final, la coexistencia de aterosclerosis acelerada causada por terapias inmunosupresoras y la falta de disponibilidad de métodos para el diagnóstico diferencial de estas dos formas de enfermedad es un obstáculo adicional para el diagnóstico. En particular en casos de vasculitis arterial coronaria aislada. Se diagnostica una vasculitis de la arteria coronaria (tabla1) en 50% de los pacientes con poliarteritis nodosa (con una incidencia notificada de 4 a 10 por millón por año) y en cerca de 20% de los pacientes con enfermedad de Kawasaki (incidencia de 2 por millón por año). Se reporta una incidencia similar de vasculitis de las arterias coronarias en pacientes con vasculitis de vasos grandes, como la arteritis de Takayasu y la arteritis de células gigantes (tasas de incidencia de 1 a 3 por millón cada una). Otras formas de vasculitis de vasos grandes como la enfermedad de Erdheim-Chester rara vez se asocian con vasculitis de las arterias coronarias (alrededor de 5% de los casos). También se reportó que la incidencia de afectación vascular en la enfermedad de Behçet está en el rango de 50%.
4. Patogenia y patología
La patogenia de la vasculitis de las arterias coronarias es compleja, multifacética, depende de la enfermedad específica y se media de forma esencial por factores extrínsecos y del huésped, como la inflamación inmunomediada y las reacciones dependientes de autoanticuerpos. Se reportó sobre citocinas inflamatorias, en parte específicas para cada afección, que incluyen interferón gamma (IFN-γ), factor de necrosis tumoral alfa (TNF-α) e interleucinas T-1. Los hallazgos histopatológicos en la autopsia también son específicos para cada condición. La vasculitis coronaria causada por poliarteritis nodosa se caracteriza por infiltración intramural, perivascular de linfocitos y macrófagos, así como necrosis fibrinoide que provocan la destrucción de la pared arterial. La arteritis de Takayasu y la arteritis de células gigantes se asocian con hiperplasia de la íntima, arteritis granulomatosa y aterosclerosis coronaria. Por el contrario, la enfermedad de Kawasaki se caracteriza por una infiltración multicelular de la pared arterial que causa necrosis de la lámina elástica interna. Como resultado de estos diferentes mecanismos, los hallazgos de inflamación crónica, tejido cicatricial, necrosis y estenosis pueden estar presentes en todas las formas de vasculitis de la arteria coronaria. Los aneurismas de la arteria coronaria son una entidad más rara como causa, como se mencionó con anterioridad, por el debilitamiento de la pared arterial que se media por una hiperactividad de metaloproteinasas y metaloelastasas. Los mismos mecanismos, junto con la liberación de sustancias proangiogenéticas de los eosinófilos y la formación resultante de capilares débiles que pueden romperse en la media arterial, pueden conducir a la disección espontánea en la arteritis eosinofílica. Las trombosis arteriales pueden resultar de la estasis sanguínea en estos aneurismas y/o en estenosis que conducen a isquemia miocárdica. Como en todas las afecciones inflamatorias, la incidencia de aterosclerosis coronaria típica también es mayor en pacientes con vasculitis. En raras ocasiones, también se describen embolias de lesiones de válvulas cardíacas de la granulomatosis de Wegener que causan oclusión de la arteria coronaria.
5. Manifestaciones clínicas y diagnóstico
Los síntomas y signos que se asocian con la afectación del sistema difieren de forma significativa entre las diferentes entidades, lo que refleja los diferentes órganos involucrados. Debido a la relativa rareza de estas enfermedades, es fácil pasar por alto un diagnóstico. La presentación clínica puede no ser específica, pero las características clínicas que podrían sugerir una etiología inflamatoria incluyen marcadores inflamatorios elevados que no se pueden explicar de otra manera, signos de inflamación como fiebre, escalofríos, sudores nocturnos, pérdida de peso, soplos subclavios o aórticos, que sugieren estenosis atípica en otros distritos, antecedentes de afectación multisistémica, como eventos isquémicos abdominales, a edad temprana, sobre todo en ausencia de predisposición genética o factores de riesgo grave. Pacientes con sospecha de vasculitis, en particular arteritis de Takayasu, deberían someterse a imagen del árbol arterial con angiografía por resonancia magnética (que tiene la ventaja de evitar la exposición a medio de contraste y radiación) o tomografía computarizada para evaluar la presencia de aneurismas, estenosis, engrosamiento de la pared arterial, y/o lesiones semejantes a masas que rodean las arterias coronarias como se revisaron de manera reciente. En particular, la tomografía computarizada proporciona información precisa no invasiva sobre las lesiones luminales y murales en la aorta y sus ramas, lo cual es importante tanto en el diagnóstico como en el seguimiento de la progresión de la enfermedad. La tomografía computarizada permite la detección y la cuantificación de estenosis y aneurismas que pueden estar presentes de manera virtual en todas las formas de vasculitis; los hallazgos que son típicos de afecciones específicas incluyen “lesiones en parches” (estenosis focales en la enfermedad de Takayasu) y lesiones de “salchichas envueltas” (anillos de atenuación de tejidos blandos que rodean las arterias coronarias) en la periarteritis coronaria. Las ecografías de otros distritos, incluidos el tórax, el abdomen, la cabeza y el cuello, pueden ser útiles, en particular para detectar engrosamientos de la pared que no pueden diagnosticarse mediante angiografía. La tomografía por emisión de positrones, a menudo en combinación con tomografía computarizada o resonancia magnética, se utiliza para el diagnóstico de vasculitis de grandes vasos. Los segmentos arteriales que presentan valores de captación estandarizados aumentados pueden ser sugestivos de enfermedad; sin embargo, estas anomalías no están presentes en las coronarias. En la angiografía, la vasculitis de la arteria coronaria puede manifestarse bajo la forma de estenosis coronaria, aneurisma (Figura 2), disección (Figura 3, más frecuente en periarteritis eosinofílica), espasmo (Figura 4) o rotura coronaria (Tabla 1). Se describió muerte súbita, angina típica, infarto agudo de miocardio, arritmias auriculares y ventriculares, alteraciones de la conducción o insuficiencia cardíaca. Aunque ningún hallazgo específico en la angiografía coronaria permite un diagnóstico seguro, las características asociadas con la enfermedad arterial coronaria atípica, avanzada o temprana pueden sugerir una etiología inflamatoria. Por ejemplo, la enfermedad de Kawasaki y la poliarteritis nodosa a menudo se asocian con aneurismas grandes. La poliarteritis nodosa a menudo presenta aneurismas multifocales con una apariencia de “cuentas en una cuerda” o nodular, que también están presentes en pacientes con vasculitis asociada a anticuerpos anticitoplasma de neutrófilos (ANCA) y enfermedad de Behçet. Las lesiones arteriales en la enfermedad de Behçet pueden ser oclusivas o aneurismáticas. La arteritis de células gigantes se asocia con lesiones coronarias grandes, y en la arteritis de Takayasu, las lesiones coronarias se clasifican en tres tipos principales: 1, estenosis u oclusión de los ostios coronarios (60-80%); 2, enfermedad difusa que puede afectar todas las ramas epicárdicas o sólo segmentos focales (10-20%); y 3, aneurismas coronarios (0-5%). La coronariografía no detecta inflamación, necrosis o engrosamiento reactivo de la pared arterial. Por lo tanto, se recomienda la evaluación no invasiva de la circulación cardíaca y extracardíaca mediante tomografía computarizada o resonancia magnética en pacientes con vasculitis de vasos grandes (arteritis de células gigantes y arteritis de Takayasu), medianos (poliarteritis nodosa) o variables (enfermedad de Behçet). Los pacientes con vasculitis de vasos pequeños (es decir, asociada a ANCA) a menudo tienen mio o pericarditis asociadas, que también deben investigarse mediante resonancia magnética. El engrosamiento de los tejidos blandos periarteriales o la compresión extrínseca también son características de afecciones más raras, como la enfermedad relacionada con la inmunoglobulina G4 (IgG4) o la enfermedad de Erdheim-Chester.
6. COVID-19 e inflamación coronaria
El impacto del COVID-19 en la biología vascular es un tema de gran atención reciente, ya que parece ser responsable de un porcentaje significativo de los resultados negativos. Se reportaron trombosis macro y microvasculares que involucran arterias, venas, arteriolas, capilares y vénulas en todos los órganos principales, junto con evidencia de endotelitis en diferentes lechos vasculares. Las lesiones presentaban endotelitis linfocítica difusa y cuerpos apoptóticos, probablemente después de la adhesión del virus a la célula y la transmisión de señales de la vía proinflamatoria y apoptótica. El estrés oxidativo, el aumento de la producción y liberación de quimiocinas, citocinas y subproductos de patrones moleculares que se asocian al daño parecen ser los responsables de la endotelitis. El reclutamiento de neutrófilos por productos de la lesión vascular está mediado por varias vías, incluidas las proteínas cinasas activadas por PI3K/AKt/eNOS/NF-Kβ y ERK1/2/P38 MAPK y conduce a una mayor inflamación mediante la liberación de TNF-α, IL-1, e IL-8, y trampas extracelulares de neutrófilos (NET). Los mecanismos que conducen a la necrosis y la apoptosis se resumieron de forma reciente en revisiones específicas. La citotoxicidad que se asocia a COVID-19 encuentra lugar principalmente dentro del sistema microcirculatorio, lo que lleva a cambios ultraestructurales y disfunción vascular. Además, el aumento de la biodisponibilidad del vasoconstrictor angiotensina II debido a la disminución de su receptor ACE-2 mediada por COVID-19, es un mecanismo reconocido de disfunción de las células endoteliales. La liberación de factores de von Willebrand que sigue al daño y la activación endoteliales puede, a su vez, reclutar y activar las plaquetas circulantes, lo que contribuye a la producción mejorada de procoagulantes, citocinas inflamatorias y NET. Puede resultar en isquemia miocárdica tipo I y II. Junto con eventos trombóticos, la vasculitis por lo tanto ocurre en COVID-19, pero su papel en los múltiples casos que se reportan de síndrome coronario agudo en pacientes con COVID-19 aún no está claro.
COVID y enfermedad similar a Kawasaki
Como se describió con anterioridad, se cree que la enfermedad de Kawasaki es el resultado de una respuesta inmune innata excesiva a los patógenos virales. Aunque el mecanismo aún no se dilucida, se propuso como vía la participación del estimulador de genes de interferón (STING, un sensor de ADN citosólico y una proteína adaptadora en la vía del IFN tipo I y del factor nuclear [NF]-κB, que conduce a la hipercoagulopatía por medio de la producción de factor tisular por macrófagos). Una situación similar puede ocurrir en las infecciones por COVID-19, donde la unión del SARS-COV-2 a ACE2 aumenta la activación de la vía STING. Las hiperrespuestas inmunes resultantes, la disminución del recuento de linfocitos, el aumento de las poblaciones de monocitos que secretan citocinas citotóxicas y el aumento de las respuestas de las células B y T configuran una enfermedad similar a Kawasaki que puede resultar en síndrome de choque tóxico o enfermedad inflamatoria multisistémica. Hasta en 25% de los niños afectados por COVID-19, se reportaron dilataciones coronarias y se publicaron varios casos de miocarditis por COVD-19. En una encuesta reciente de 149 pacientes, los niños con “síndrome de Kawacovid” eran mayores de forma significativa y presentaban con mayor frecuencia afectación gastrointestinal y respiratoria. La afectación cardíaca en forma de miocarditis fue más común (60%) como en Kawasaki tradicional, pero las anomalías de las arterias coronarias fueron raras. Cerca de 40% de los pacientes con Kawacovid presentaron hipotensión/choque no cardiogénico. En la resonancia magnética cardíaca, se reportó evidencia de edema miocárdico difuso. La presentación clínica del síndrome inflamatorio multisistémico resultante en niños (MIS-C) refleja la de Kawasaki, pero existen diferencias entre los dos con respecto a la edad (desde la primera infancia hasta la adolescencia tardía en comparación con la primera infancia en Kawasaki); linfopenia y trombocitopenia más frecuentes, estrés ventricular cardíaco como miocarditis y coagulopatía en MIS-C, afectación gastrointestinal más frecuente, miocarditis que conduce a choque cardiogénico y un perfil de citocinas diferente (IL-6 e IL-8 en MIS-C, IL- 1 en Kawasaki).
7. Procedimientos de revascularización en vasculitis coronaria
7.1. Síndromes coronarios crónicos
Todos los pacientes con vasculitis conocida deben tratarse con ácido acetilsalicílico. En cuanto a la indicación de revascularización, los métodos tradicionales de valoración de la vitalidad (ecocardiografía de estrés con dobutamina a dosis baja, gammagrafía, resonancia magnética) e isquemia (electrocardiografía en ejercicio, ecocardiografía de estrés o gammagrafía, etc.) mantienen su vigencia. Hay pocos datos, de naturaleza anecdótica, sobre los resultados de las intervenciones en pacientes con arteritis coronaria; las pautas actuales de la Asociación Americana del Corazón recomiendan intervenciones percutáneas en pacientes con afectación de un solo vaso o enfermedad focal multivaso. Sin embargo, es posible que las intervenciones coronarias percutáneas no proporcionen una revascularización adecuada en casos de enfermedad difusa. Dada la gravedad de estas enfermedades y su rareza, no hay ningún ensayo prospectivo aleatorizado para determinar los resultados de los diferentes métodos de revascularización (cirugía intervencionista versus cirugía de derivación). En pacientes con arteritis de Takayasu, se reportó una tasa mayor de fracaso de la lesión diana después de la intervención coronaria percutánea en comparación con la cirugía (razón de momios 7.4 [2.4-23.1], p = 0.01), por lo que se recomienda la cirugía en estos pacientes tras la inducción de la inmunosupresión. La afectación de la aorta ascendente es un factor claro que complica la cirugía de injerto de derivación arterial libre y venosa. Además, la arteria mamaria interna también puede ser un objetivo de enfermedad en pacientes con arteritis de Takayasu, lo que complica el injerto de derivación. La cicatrización de la herida puede comprometerse o ser más lenta en pacientes con enfermedad activa. Por último, los aneurismas de las arterias coronarias se pueden ocluir al enrollar o implantar injerto de stents cubiertos, aunque el riesgo de trombosis y reestenosis continúa elevado. La resección de aneurisma/trombectomía y la cirugía de derivación también son opciones.
7.2. Síndromes coronarios agudos
Los pacientes con aneurismas coronarios tienen un riesgo elevado de infarto de miocardio. Dada la corta edad, la terapia intervencionista en este entorno a menudo no es factible. En caso de trombosis grave que se produzca dentro del aneurisma, también se reportó de tratamiento con heparina, antagonistas de IIb IIIa y fibrinolíticos en la edad pediátrica. Sin embargo, se desconocen los resultados de estos procedimientos. De forma independiente del tipo de revascularización, se debe aplicar terapia antiplaquetaria y se recomienda la terapia antiplaquetaria de por vida en todos los pacientes con aneurismas coronarios. Los datos sobre anticoagulación en pacientes con aneurismas también son muy escasos, y algunos autores desaconsejan este tipo de abordaje por temor a la progresión de los aneurismas mientras que otros reportan una incidencia reducida de infartos agudos de miocardio en pacientes tratados con una combinación de aspirina y warfarina. Además, a pesar de la anticoagulación, la trombosis en aneurismas gigantes puede ocurrir debido a la estasis sanguínea y la disminución de la tensión tangencial de la pared. Por último, la inmunosupresión es un pilar de la terapia, y se reportan casos de regresión “espontánea” de estenosis de las arterias coronarias. Se puede considerar el trasplante cardíaco en pacientes que no se consideren aptos para la revascularización.
8. Conclusiones
La vasculitis de las arterias coronarias es rara, pero es una de las causas más frecuentes de enfermedad de las arterias coronarias en pacientes jóvenes. Sus manifestaciones anatómicas pueden incluir estenosis de arterias coronarias, aneurismas, trombosis y disección espontánea; y sus consecuencias pueden ser graves. Aunque es malo el pronóstico de la vasculitis coronaria, el diagnóstico y la terapia tempranos mejoran las tasas de supervivencia. Tanto los métodos invasivos como los no invasivos proporcionan información esencial en el diagnóstico. Ahora son necesarios estudios a gran escala para investigar más a fondo la incidencia, el rendimiento diagnóstico y la terapia de este grupo de enfermedades poco común y heterogéneo.
Coronary Vasculitis

Centro Regional de Alergia e Inmunología Clínica CRAIC, Hospital Universitario “Dr. José Eleuterio González” UANL, Monterrey, México
Dra. Med. Sandra Nora González Díaz Jefe y Profesor
Dra. Marisela Hernández Robles Profesor
Dr. Jesús Eduardo Uc Rosado Residente 1er Año
Dra. Alejandra Macías Weinmann Profesor




No hay comentarios:
Publicar un comentario
Nota: solo los miembros de este blog pueden publicar comentarios.