
Caso clínico Una mujer de 66 años con antecedentes de hipertensión, diabetes e hiperlipidemia acude al servicio de urgencias por malestar general, fiebre y un exantema que se ha desarrollado en el transcurso de horas. La erupción es macular e involucra las membranas mucosas. Afirma que está empezando a tener dificultad para tragar debido al dolor. Cuando se aplica presión mecánica en el borde de las ampollas o en la piel normal, se producen ulceraciones adicionales. Ella informa que actualmente está completando un ciclo de antibióticos para una ITU que le diagnosticaron la semana pasada. La presión arterial es 167/89, la FC es 104, la FR es 14, la temperatura es 37,2°C. |
SSJ/NET
El síndrome de Stevens-Johnson (SSJ) es una afección cutánea potencialmente mortal que se manifiesta en un espectro de gravedad. Comienza con un pródromo de fiebre alta, síntomas parecidos a la gripe, sensibilidad en la piel y ampollas.
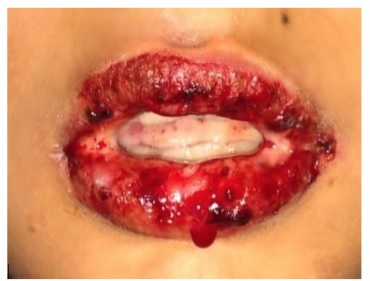
El exantema característico se describe comúnmente como un exantema macular eritematoso confluente con centros purpúricos que ampollan y pelan. El SSJ involucra al menos el 10% del área de superficie corporal; la necrólisis epidérmica tóxica (NET) se diagnostica si la erupción involucra al menos un 30% de la superficie corporal.
El compromiso característico de la mucosa puede provocar síntomas como picazón conjuntival y dolor al tragar. La erupción generalmente comienza en el torso y la cara y se extiende al resto del cuerpo, por lo general no afecta las palmas y las plantas. Esta erupción tiene un signo de Nikolsky positivo, en el que la presión lateral sobre la piel intacta provoca el desprendimiento de la epidermis.
Esta es un rasgo distintivo importante para diferenciar SSJ/NET de las otras erupciones potencialmente mortales que se comentan más adelante. De estos pacientes, del 70 al 100% tienen afectación de la mucosa y hasta el 80% tienen afectación ocular que cursa con conjuntivitis y secreción. Ante este hecho, es importante realizar un examen ocular y ginecológico minucioso.
Los factores de riesgo para desarrollar SSJ/NET incluyen pacientes con cáncer activo, VIH, mujeres y que reciben muchos medicamentos. Los medicamentos más comúnmente involucrados incluyen trimetoprima-sulfametoxazol, fenobarbital, carbamazepina, lamotrigina, paracetamol y agentes quimioterapéuticos.
Las penicilinas se citan como el agente antibiótico que más comúnmente causa SSJ/NET. No está claro si esto se debe a la frecuencia con la que se recetan penicilinas o si el antibiótico se estaba usando para tratar una manifestación cutánea temprana del SSJ como una infección bacteriana. Por lo tanto, se debe realizar una cuidadosa historia y cronología para evaluar el verdadero detonante.
El pilar del tratamiento SSJ/NET es el tratamiento de sostén.
Primero, discontinúe el agente causante. El cuidado local de las heridas, el control del dolor y la administración de líquidos por vía intravenosa deben continuarse durante el curso de la enfermedad, de forma muy similar a como lo haría con un paciente quemado.
Un análisis retrospectivo en 2010 recomendó un volumen de líquido intravenoso inicial de 2 ml/kg de peso corporal multiplicado por el % de superficie corporal con desprendimiento epidérmico en las primeras 24 horas.
Tienen una alta sospecha clínica de infecciones secundarias que se deben más comúnmente a S. aureus y Pseudomonas aeruginosa. Aunque no se recomiendan antibióticos profilácticos, los cultivos de piel pueden ayudar a guiar la terapia.
Los estudios han demostrado que, lamentablemente, no hubo beneficio en la supervivencia para los tratados con corticoides sistémicos en comparación con los tratados con la atención habitual.
Una herramienta de decisión útil para determinar el entorno clínico más adecuado para el tratamiento de su paciente es la escala SCORTEN (que se ve a continuación), que puede evaluar la gravedad de la enfermedad; a aquellos con una puntuación mayor o igual a 2 se recomienda su internación en UCI o unidad de quemados.
| Factor pronóstico | Puntos |
| Edad > 40 años | 1 |
| Taquicardia > 120 lpm | 1 |
| Neoplasia | 1 |
| Desprendimiento inicial > 10% | 1 |
| Urea > 60 mg/dL | 1 |
| Bicarbonato sérico < 20 mEq/L | 1 |
| Glucosa en sangre > 250 mg/dL | 1 |
| SCORTEN | % Mortalidad |
| 0-1 | 3 |
| 2 | 12 |
| 3 | 35 |
| 4 | 58 |
| > 4 | 90 |
Como el SSJ/NET tiene una amplia gama de presentaciones y una tasa de mortalidad potencialmente alta, es importante distinguir de otras erupciones que se presentan de manera similar. De hecho, cuando se sugirió por primera vez el término NET en 1956, el documento incluía pacientes con lo que ahora se ha diferenciado en erupciones fijas por medicamentos y síndrome estafilocócico de la piel escaldada (SEPE). A continuación, se discuten varios “imitadores” de SSJ/NET que tienen diferentes pronósticos, causas y vías de manejo.
> Pustulosis exantematosa aguda generalizada
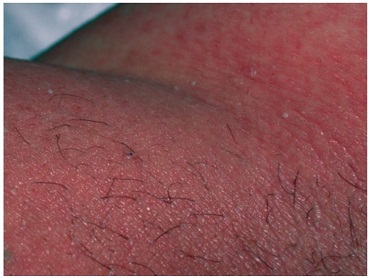
La pustulosis exantematosa aguda generalizada (PEAG) es una reacción cutánea poco común pero grave que a menudo se confunde con SSJ/NET. Es una reacción farmacológica que se presenta con pústulas estériles no foliculares sobre una base eritematosa y edematosa. Es más común en superficies de flexión y a menudo se ve primero en áreas intertriginosas que se extienden al tronco.
Por lo general, se desarrolla rápidamente y se presenta dentro de las 24 a 48 horas posteriores al inicio de un medicamento, a diferencia del SSJ, que puede comenzar días o semanas después de tomar un medicamento. La afectación de la mucosa oral se observa en el 25%, a diferencia de la participación de la mayoría de las personas con SSJ.
Muchos medicamentos pueden causar esta reacción, en particular penicilinas, quinolonas, sulfonamidas e hidroxicloroquina. A menudo se observan fiebre, leucocitosis, elevación de los reactantes de fase aguda y eosinofilia. La PEAG suele ser un diagnóstico clínico, pero la biopsia puede confirmarlo. Además, después de la resolución, una prueba de parche puede ser útil para determinar el agente causal.
Convenientemente, el manejo es muy similar al SSJ/NET, que consiste principalmente en suspender el agente causante y cuidados de sostén que generalmente conducen a la resolución del exantema en 2 semanas. Los esteroides tópicos son útiles, pero nuevamente los esteroides sistémicos no tienen un beneficio claro.
> Eritema multiforme
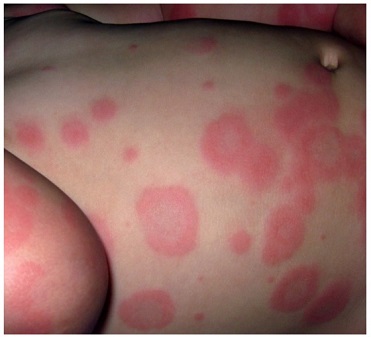
El eritema multiforme (EM) es una afección inmunomediada que se presenta con lesiones diana distintivas con o sin afectación de la mucosa.
Las lesiones diana son clásicas, pero la erupción tiene una presentación variada que evoluciona a lo largo de la enfermedad. Una distinción significativa es que las lesiones de EM tienden a ser papulares en oposición a las lesiones diana atípicas de SSJ que tienden a ser de naturaleza macular.
Cuando estas lesiones afectan áreas de las mucosas, generalmente se denomina eritema multiforme mayor. Cuando hay poca o ninguna afectación de la mucosa, se puede describir como eritema multiforme menor. Las lesiones se desarrollan típicamente en 3 a 5 días y se resuelven en 2 semanas.
Existe una variedad de factores de riesgo que contribuyen al desarrollo de EM, que incluyen, entre otros, infecciones, fármacos, neoplasias malignas y enfermedades autoinmunes. Sin embargo, las infecciones representan aproximadamente el 90% de los casos, siendo el virus herpes simple (VHS) más común en adultos y el micoplasma en niños.
El tratamiento del eritema multiforme es típicamente un cuidado sintomático, con esteroides tópicos y antihistamínicos. Si hay afectación de las mucosas, se deben considerar los esteroides intravenosos u orales.
La afectación grave de las membranas mucosas, como ocurre con el multiforme mayor, puede requerir hospitalización y se debe considerar una consulta con el oftalmólogo si hay afectación ocular. Es importante señalar que el tratamiento de una infección causante subyacente no afecta la gravedad o la duración de la EM.
> Reacción farmacológica con eosinofilia y síntomas sistémicos (DRESS)
Como indica el nombre, es otra reacción grave inducida por fármacos que puede ser difícil de diferenciar.
Este síndrome cursa con fiebre, eosinofilia o elevación de linfocitos atípicos, linfadenopatía, edema facial y malestar generalizado.
Esta erupción generalmente no se presenta hasta 2 a 8 semanas después de la exposición al fármaco y puede continuar o incluso empeorar después de la retirada del agente causal. Incluya función hepática al ordenar los estudios de laboratorio, ya que generalmente hay afectación de órganos; con mayor frecuencia el hígado, los pulmones o los riñones.
Algunos de los fármacos más comunes que causan DRESS incluyen anticonvulsivantes aromáticos (por ejemplo, fenitoína, carbamazepina y fenobarbital), sulfonamidas, alopurinol y vancomicina. El DRESS generalmente se presenta con una erupción morbiliforme que comienza en el tronco y las extremidades superiores.
La erupción se vuelve edematosa provocando edema periorbitario en la cara. Puede tomar muchas formas y presentarse con pústulas, foliculitis o descamación o incluso una forma rara con afectación de las mucosas. Por tanto, puede ser muy difícil de diagnosticar y diferenciar de otras erupciones y requiere un alto índice de sospecha.
El tratamiento implica esteroides tópicos. Los esteroides sistémicos con una disminución gradual durante 6 a 8 semanas están indicados para pacientes con signos sistémicos como hepatitis, pleuritis, neumonía y daño renal. Para los casos refractarios se han utilizado IgIV y plasmaféresis.
> Pénfigo vulgar
El pénfigo vulgar es una enfermedad autoinmune crónica que se caracteriza por acantólisis. La destrucción de los desmosomas en la epidermis da como resultado la pérdida de la adhesión entre los queratinocitos, lo que da como resultado ampollas dolorosas. Estas ampollas son signo de Nikolsky positivo.
Las distinciones importantes de esta erupción de las otras que discutidas previamente es que el prurito generalmente está ausente y casi siempre hay afectación de la mucosa, principalmente de la mucosa bucal.
Los factores de riesgo incluyen luz ultravioleta y medicamentos. Ciertos fármacos tiol, como la penicilamina y el captopril, son los agentes causales más comunes. Los glucocorticoides sistémicos son el tratamiento de elección y son responsables del control rápido de esta enfermedad.
También hay estudios emergentes que investigan el uso de rituximab, un anticuerpo monoclonal o combinaciones de medicamentos inmunomoduladores sistémicos no esteroideos, como azatioprina o micofenolato como complemento de los glucocorticoides con resultados positivos.
El uso de estos complementos reduce la cantidad de esteroides necesarios y, por lo tanto, produce menos eventos adversos. Para casos refractarios severos, la inmunoglobulina IV también puede ser beneficiosa. En estos casos, consultar a un reumatólogo o dermatólogo.
> Meningococcemia
Uno de los primeros síntomas en presentarse de la meningitis es una erupción clásica que puede progresar de inespecífica a petequial y a hemorrágica en cuestión de horas.
El exantema comienza como petequias de 1-2 mm en el tronco y las extremidades inferiores. Aproximadamente el 50% de los pacientes con meningococcemia presentarán petequias.
El grado de trombocitopenia puede estimarse por la presencia de estas petequias y, por lo tanto, aumenta la preocupación por posibles complicaciones hemorrágicas secundarias a la CID. Estas petequias pueden fusionarse en una púrpura más grande. La púrpura fulminante es una complicación grave que puede ocurrir en aproximadamente el 15-25% de las personas con meningococcemia diagnosticada.
Se caracteriza por el inicio agudo de hemorragia cutánea, CID y trombosis vascular. Se forman ampollas y vesículas que eventualmente pueden conducir a una necrosis gangrenosa. A diferencia de muchas de las erupciones que hemos comentado hasta ahora, la erupción por meningococcemia es un síntoma de un proceso patológico mucho más extenso que la enfermedad en sí.
> Síndrome estafilocócico de la piel escaldada (SEPE)
El síndrome de piel escaldada por estafilococos es una afección cutánea potencialmente mortal causada por una toxina bacteriana de Staphylococcus aureus que sufre diseminación hematógena desde la piel.
Se presenta con piel eritematosa dolorosa que comienza principalmente en áreas de alta fricción como los pliegues cutáneos. Bullas flácidas, descamación y un signo de Nikolsky positivo caracterizan la erupción.
Es importante destacar que, a diferencia de SSJ/NET, no hay afectación de la mucosa y la erupción es más superficial. Esta erupción afecta con mayor frecuencia a bebés y niños y puede presentarse temprano con irritabilidad y una ingesta oral deficiente. Si se observa en adultos, la tasa de mortalidad es alta, a menudo hasta el 60% debido a la alta carga de infección, mientras que la tasa de mortalidad en los lactantes es del 5%.
El tratamiento es de sostén, con antibióticos que cubran la infección por estafilococos. La nafcilina, la oxacilina y la vancomicina son opciones comunes, ya que la mayoría de las bacterias que causan SEPE son resistentes a la penicilina. Los esteroides están contraindicados debido a la inmunosupresión.
> Eritrodermia
La eritrodermia, también llamada dermatitis exfoliativa, es una erupción poco común que se identifica por una descamación generalizada que cubre la mayoría de las superficies de la piel.
Se ve con mayor frecuencia en la población masculina de edad avanzada. Muchas causas pueden provocar la erupción, incluidas afecciones cutáneas subyacentes, reacciones a fármacos, HIV y linfoma cutáneo de células T. La piel está enrojecida, caliente, pruriginosa y dolorosa.
El paciente a menudo tiembla debido a la pérdida de calor por vasodilatación cutánea. Puede haber otros hallazgos del examen que se correlacionen con la causa subyacente, como psoriasis ungueal; en los linfomas de células T pueden observarse linfadenopatía y esplenomegalia.
La erupción tiene un inicio rápido si es causada por fármacos, mientras que puede tardar más en desarrollarse cuando se debe a otras etiologías. El tratamiento consiste en interrumpir los posibles medicamentos causales, cuidados de sostén y esteroides tópicos.

Puntos clave 1) Haga una historia clínica completa. Identificar cualquier exposición a nuevos fármacos durante los últimos meses es una información valiosa. 2) El signo de Nikolsky (desprendimiento de la piel con presión lateral) y la presencia de afectación de la mucosa pueden ser factores diferenciadores importantes en la identificación de erupciones críticas: SSJ/NET, pénfigo vulgar y SEPE son Nikolsky positivos. 3) Tratamiento
|




No hay comentarios:
Publicar un comentario
Nota: solo los miembros de este blog pueden publicar comentarios.