
Hicieron falta casi 100 años para darse cuenta de que el lupus eritematoso, que se pensó de manera inicial que era una entidad de la piel, es una enfermedad sistémica que no perdona ningún órgano y que involucra una respuesta autoinmune aberrante en su patogenia. La participación de órganos y tejidos vitales como el cerebro, la sangre y el riñón en la mayoría de los pacientes, la gran mayoría de los cuales son mujeres en edad fértil, impulsa los esfuerzos para desarrollar herramientas de diagnóstico y terapias eficaces. La prevalencia varía de 20 a 150 casos por 100.000 personas y parece estar en incremento a medida que la enfermedad se reconoce de manera más fácil y mejoran las tasas de supervivencia. En los Estados Unidos, las personas de ascendencia africana, hispana o asiática, en comparación con las de otros grupos raciales o étnicos, tienden a tener una mayor prevalencia de lupus eritematoso sistémico (LES) y una mayor participación de órganos vitales.
La tasa de supervivencia a 10 años se incrementó de manera significativa en los últimos 50 años a más de 70%, debido a una mayor conciencia de la enfermedad, el uso extenso y más sabio de medicamentos inmunosupresores y un tratamiento más eficiente de las infecciones, la principal causa de muerte.
Aunque son necesarios niveles bajos de autorreactividad y autoinmunidad para la selección de linfocitos y, en general, para la regulación del sistema inmunológico, en ciertos individuos, la autoinmunidad avanza mediante múltiples vías y conduce a inflamación y daño de órganos. Los diversos mecanismos no contribuyen de la misma forma a la expresión de la enfermedad en todos los pacientes con LES, como se comentan a continuación. Parece que la heterogeneidad clínica de la enfermedad se acompaña de múltiples procesos patogénicos, lo que justifica el llamado al desarrollo de la medicina personalizada (Fig. 1).
Genes y genética
Durante las últimas dos décadas, estudios extensos de asociación y metaanálisis de todo el genoma identificaron cerca de 150 nuevos loci de riesgo de LES en múltiples ascendencias. El uso extensivo de la secuenciación del exoma reveló un número creciente de casos monogénicos de LES. De forma interesante, varios de los loci de riesgo abarcan múltiples enfermedades autoinmunes que estipulan enfermedades comunes. Los estudios funcionales de loci compartidos pueden ayudar a reclasificar las enfermedades autoinmunes según las vías compartidas, junto con las características clínicas. Se publicaron listas detalladas de variantes genéticas y en la Tabla 1 se enumera un número limitado, junto con pruebas que apoyan la participación en la patogenia de la enfermedad. Varias variantes se relacionaron a nivel genético, algunas con biología de apoyo, a mecanismos patogénicos y manifestaciones clínicas específicas, lo que apunta de manera fuerte a la heterogeneidad de la enfermedad. Varios genes relacionados con la respuesta inmune se encuentran regulados por medio de interacciones de cromatina a larga distancia. Aún faltan estudios que aborden las interacciones a larga distancia entre variantes de genes en el LES, pero, con el advenimiento de nuevas tecnologías, surgirán tales estudios.
Se necesita una mejor comprensión del epigenoma para comprender cómo la contribución genética complementa a la enfermedad. Se reconoció la disminución de la metilación del ADN de ciertos genes en las células T del LES y se atribuyeron a una función deficiente de la enzima de remetilación de CpG DNMT1. Por ejemplo, la hipometilación de TNFSF5, que se encuentra en el cromosoma X, produce un aumento de la expresión del ligando CD40 (CD40L) en las células T de las mujeres, y la hipometilación de Il10 aumenta la producción de la interleucina (IL)-10. Sin embargo, la metilación de genes en el LES es más complicada y no sigue un patrón unidireccional. Por ejemplo, el locus CD8 se encuentra metilado en linfocitos T de pacientes con LES, lo que da lugar a la generación de linfocitos T CD3+CD4−CD8–, mientras que el locus Il2 se encuentra hipermetilado, lo que da como resultado una producción baja de IL-2. La administración de un agente desmetilante de manera específica a células CD4+ o CD8+ en ratones propensos al lupus suprime la expresión de la enfermedad al mejorar la expresión de FoxP3 y mantener la expresión de CD8.
En las células T del LES, el modificador del elemento de respuesta de AMP cíclico (CREMα) se une a varios elementos reguladores dentro del grupo CD8 y recluta modificadores de histonas, incluidos DNMT3a y la histona metiltransferasa G9a, lo que provoca un silenciamiento estable de CD8A y CD8B. Un proceso similar tiene lugar en el locus Il2, lo que resulta en una disminución de la producción de IL-2. Por el contrario, el reclutamiento de STAT3 a las regiones reguladoras Il10 media el reclutamiento de la histona acetiltransferasa p300, lo que da como resultado una mayor expresión génica.
Se sospecha que un gran número de microARN controla al menos un tercio de la estabilidad y traducción del ARNm humano y, y de manera razonable, se estudiaron en el LES. En la Tabla 2 se presenta una lista limitada. Las concentraciones séricas de microARN pueden servir como biomarcadores de enfermedades, y el desarrollo de antagonistas, usados para silenciar micoARN endógenos, puede ayudar a controlar la enfermedad.
La contribución de las modificaciones epigenéticas a la expresión de la enfermedad complementa la susceptibilidad genética. Una mejor comprensión de los procesos bioquímicos implicados debería ofrecer oportunidades únicas para desarrollar terapias nuevas y de forma personalizada.
De manera aproximada un tercio de los gemelos monocigóticos son concordantes de manera clínica con el LES, lo que indica la importancia de los factores ambientales en la expresión de la enfermedad. El entorno, como la luz ultravioleta (UV), y la reactividad cruzada entre autoantígenos y moléculas definidas por virus y otros patógenos es importante en la patogénesis del LES. Los microbios, como los organismos comensales inocuos que colonizan el intestino, la piel, las cavidades nasales y la vagina, pueden desencadenar y mantener la inflamación autoinmune en huéspedes susceptibles de manera genética. Se demostró de manera amplia que la microbiota modifica la respuesta inmune, incluido el desarrollo de células T cooperadoras tipo 1 (TH1), TH2 y T reguladoras (Treg) y se implicó en varias enfermedades autoinmunes.
Se afirmó desde hace mucho tiempo que la reactividad cruzada entre especies bacterianas y autoantígenos contribuye a la expresión de enfermedades en individuos susceptibles. La bacteria Propionibacterium propionicum, que codifica un ortólogo de la proteína de unión a ARN Ro60, se encontró en lesiones cutáneas de pacientes con lupus eritematoso cutáneo y se demostró que estimula las células T de memoria de pacientes con LES, lo que sugiere una participación directa de patógenos en la proliferación de células T y la producción de autoanticuerpos.
Las bacterias filamentosas segmentadas inducen células TH17 productoras de IL-17 intestinal, y la microbiota intestinal impulsa la artritis autoinmune al promover la generación de células T cooperadoras foliculares (TFH). La microbiota de manera aparente usa la transmisión de señales del receptor tipo toll (TLR) debido a que los ratones deficientes en TNFAIP3 (A20), que desarrollan autoinmunidad, no lo hacen cuando MyD88 (una molécula central de transmisión de señales de TLR) se elimina de manera genética o los ratones se tratan con antibióticos. La microbiota se transloca desde el intestino a los ganglios linfáticos mesentéricos, el bazo y el hígado e induce células TH17 y TFH, así como vías inmunes innatas, incluido el eje del interferón tipo I (IFNα/β) de células dendríticas plasmocitoides (CDp). De manera interesante, se puede encontrar cierta microbiota en el hígado de pacientes con LES o hepatitis autoinmune. La microbiota contribuye a la expresión de la enfermedad mediante varios mecanismos, que incluyen el mimetismo molecular, la participación de la respuesta inmunitaria innata y la propagación de células TH17 proinflamatorias. En consecuencia, una mejor comprensión del papel de la microbiota en la expresión del LES debería revelar enfoques simples para controlar la autoinmunidad mediante cambios en la dieta o al cambiar la distribución de la microbiota en el intestino y en otros lugares.
Sexo
A pesar de que más de 90% de las personas afectadas por LES son mujeres, todavía no se tiene una comprensión clara de los mecanismos causales. Es bien sabido que las personas con XXX (síndrome de Klinefelter) son propensas al LES y que los cambios epigenéticos en ciertos genes patógenos (por ejemplo, TNFSF5, que codifica CD40L) contribuyen a la expresión de la enfermedad. Además, seis loci de susceptibilidad al LES se asignan al cromosoma X, cuatro de los cuales (TLR7, TMEM187, IRAK1 [cinasa que interactúa con MyD88] y el CXorf21 inducible por IFN-α) pueden escapar de la inactivación del cromosoma X.
El estrógeno altera los umbrales de apoptosis y activación de células B, y el receptor α de estrógeno contribuye a la inflamación autoinmune mediada por células T al promover la activación de las células T, y también promueve el lupus en ratones NZB × NZW F1. A nivel molecular, el estrógeno favorece la expresión de CREMα, que se sabe que controla la expresión de Il2 e Il17. El análisis de expresión génica reveló una red relacionada con la autoinmunidad con sesgo femenino impulsada por el factor de transcripción VGLL3 que se encuentra relacionado con enfermedades autoinmunes, como el LES, el síndrome de Sjögren y la esclerodermia. Si bien todavía no se entiende por qué las mujeres representan la gran mayoría de las personas con LES, la evidencia nueva apunta a distintos procesos moleculares y la probable activación de genes definidos por el cromosoma X que se vinculan de manera genética al LES.
Alteraciones de las células inmunitarias innatas
Los factores genéticos y epigenéticos contribuyen de manera directa a alterar las células de las respuestas inmunes tanto innatas como adaptativas. Es probable que determinadas aberraciones inmunitarias provoquen otras. Los estudios en ratones y humanos son limitados debido a los enfoques reduccionistas que se necesitan para comprender la contribución de cada anomalía a la expresión de la enfermedad. Se requieren enfoques de inteligencia artificial para comprender la secuencia de eventos en cualquier paciente. Dicho conocimiento es necesario para la aplicación de protocolos de tratamiento de precisión.
Los neutrófilos en pacientes con LES muestran una mayor capacidad para formar trampas extracelulares de neutrófilos (NETosis) que albergan autoantígenos, como cromatina, ADNds y proteínas granulares. En pacientes con LES, las NETs se eliminan poco y estimulan las CDp para que produzcan IFN tipo I mediante la estimulación con TLR9. La endotelina 1 y el factor 1α inducible por hipoxia parecen mediar la expresión de la proteína de respuesta al estrés REDD1, que impulsa la formación de NET en el LES. Las NETs se encuentran rodeadas con factor tisular e IL-17 y son abundantes en las lesiones cutáneas discoides y en los riñones de pacientes con LES. Además, los neutrófilos esplénicos localizados en la zona perimarginal pueden inducir el cambio de clase de inmunoglobulina (Ig), la hipermutación somática y la producción de anticuerpos al activar las células B de la zona marginal. De manera interesante, los pacientes neutropénicos tienen menos células B mutadas de la zona marginal y menos Ig preinmune específica para antígenos independientes de T, lo que sugiere que los neutrófilos generan un nivel adicional de inmunidad innata en la defensa antibacteriana.
Las células dendríticas (CD) vinculan las respuestas inmunitarias innatas y adaptativas y se identificaron en la expresión del LES, ya que su activación incontrolada puede impulsar la autoinmunidad. Aunque los números disminuyen en la periferia, se encuentran activados en los tejidos inflamados, produciendo citocinas inflamatorias y ayudando a las células T y B. Los inmunocomplejos que contienen ARN inducen la expresión del ligando OX40 por las CD convencionales del LES. De manera posterior, impulsan la diferenciación de células T CD4+ vírgenes y de memoria en células TFH, que pueden ayudar a las células B y deteriorar la función de Treg. Las CD convencionales en el LES instruyen la diferenciación de plasmablastos IgG e IgA y contribuyen a la formación de estructuras linfoides ectópicas.
Las CDp se distinguen de las convencionales por la morfología y los marcadores de superficie celular y son igual de bajas en la periferia, de manera probable porque se alojan en áreas inflamadas. Activadas por agonistas de TLR7/9, producen IFN tipo I para contribuir a la expresión de la enfermedad, y la duplicación de TLR7 promueve la enfermedad. La disminución específica de CDp en ratones reduce las manifestaciones de enfermedades como la producción de autoanticuerpos, glomerulonefritis y expresión de genes inducibles por IFN. A nivel clínico, el direccionamiento de CDp con un anticuerpo BDCA2 mejora la enfermedad de la piel en pacientes con LES, mientras que la activación crónica de CDp por medio de TLR7 y TLR9 hace que las CDp sean resistentes a la inhibición de la vía NF-κB y conduce a la resistencia a los esteroides.
Los macrófagos de la zona marginal que rodean los folículos esplénicos son cruciales para la eliminación eficaz de las células apoptóticas y para la inducción de tolerancia a los autoantígenos. La fagocitosis de células apoptóticas por macrófagos de la zona marginal esplénica requiere la vía mecanosensora mediada por el coactivador transcripcional de leucemia 1 megacarioblástica. La producción de IFN tipo I por los macrófagos en respuesta al compromiso de TLR7 se habilita por el receptor TREML4 expresado en las células mieloides, y los macrófagos de los ratones Treml4-/- son hiporrespondedores a los agonistas de TLR7, mientras que los ratones propensos al lupus MRL-lpr deficientes en TREML4 muestran una autoinmunidad disminuida y nefritis. También debe tenerse en cuenta que el IFN tipo I y el factor de necrosis tumoral (TNF) cooperan para promover una firma inflamatoria en los monocitos, y dicha cooperación también se produce en los monocitos de pacientes con LES.
Se demostró que el IFN de tipo I, que afecta a múltiples componentes del sistema inmunológico, contribuye a la patogenia del LES en adultos y niños y refleja la actividad de la enfermedad. Sin embargo, el IFN tipo I solo puede no ser suficiente para causar la expresión de la enfermedad y, en algunas cepas múridas, incluso puede ser beneficioso.
El procesamiento adecuado del material apoptótico implica la activación del receptor de hidrocarburos arilo (AhR) del factor de transcripción después del compromiso de TLR9, lo que conduce a una serie de eventos que suprimen la inflamación, incluida la producción de IL-10. La eliminación de AhR en células mieloides causa autoinmunidad y su firma de transcripción se correlaciona con la actividad de la enfermedad en ratones y humanos. Esta observación puede explicar por qué TLR9 tiene un efecto protector contra la autoinmunidad.
Las plaquetas se activan en pacientes y ratones con LES mediante una serie de mecanismos, incluida la acción de los complejos inmunes y el contacto con las células endoteliales lesionadas, y muestran una firma de IFN tipo I. Una vez activadas, las plaquetas expresan y liberan CD40L y modulan la inmunidad adaptativa al activar las células presentadoras de antígenos, como las CD. Las plaquetas interactúan con las CDp en los pacientes para aumentar la secreción de IFN tipo I al activar TLR9 y TLR7. La comprensión de la contribución de las plaquetas puede revelar herramientas adyuvantes para el tratamiento del LES..
La IgE autorreactiva hace que los basófilos se alojen en los ganglios linfáticos, promueve la diferenciación de las células TH2 y mejora la producción de anticuerpos autorreactivos que causan nefritis similar al lupus en ratones que carecen de la proteína tirosina cinasa Lyn. Los pacientes con LES con concentraciones elevadas de IgE autorreactiva y basófilos activados tienen mayor actividad de la enfermedad y nefritis lúpica activa. Los estudios en ratones demostraron, de manera definitiva, el papel de los neutrófilos, los basófilos, las CDp, la activación de TLR y la producción de IFN tipo I en la expresión del LES. Varios de estos contribuyen de manera directa al daño orgánico, mientras que otros instruyen, de manera directa o indirecta, las aberraciones de la respuesta inmune adaptativa. La diversidad de las vías implicadas subraya el espectro clínico amplio de la enfermedad y es muy posible que cada elemento celular contribuya a la expresión de la enfermedad, en diversos grados..
Alteraciones de los linfocitos en el LES
Las células B en el LES se revisaron de manera extensa. La pérdida de tolerancia de las células B en distintos puntos de control explica la producción de autoanticuerpos. Los estudios de secuenciación del receptor de antígeno de células B (BCR) en niños con LES sugirieron que los defectos en distintos puntos de control en el desarrollo temprano de las células B explican la producción de autoanticuerpos. Aunque la autoinmunidad es el resultado de la falla de los puntos de control de tolerancia, existe evidencia de que puede surgir de la expansión de las células autorreactivas existentes.
Se detectaron células B asociadas a la edad (que incluyen células B IgD-CD27- y CD21lo) en trastornos autoinmunitarios humanos, incluido el LES. Su expansión se controla por el factor de transcripción IRF5, cuyas variantes se encuentran vinculadas al LES, por medio de la expresión de IL-21 y una remodelación única del paisaje. En ratones, las células B asociadas a la edad se expanden en ausencia de proteínas reguladoras de GTPasa (DEF6 y SWAP70), y se identificó que las variantes de DEF6 confieren una mayor susceptibilidad al LES.
La firma molecular de LES en las células B parece establecerse durante la fase de reposo sin tratamiento y es dominada por el enriquecimiento de motivos de cromatina accesibles para los factores de transcripción AP-1 y EGR, que se facilita, de manera probable entre otros factores, por IFN-γ. Se sabe que las células B que carecen de IgD y CD27 se expanden en pacientes con LES y producen autoanticuerpos. Estas células son hipersensibles a los agonistas de TLR7 y a la IL-21, carecen del regulador de TLR TRAF5 y tienen características de células B vírgenes recién activadas.
Aunque las reacciones alérgicas no son más frecuentes en personas con LES en comparación con sujetos normales, los anticuerpos IgE específicos para ADNds se encuentran presentes en el suero de pacientes con LES. Estos anticuerpos IgE se unen al receptor FcγRI de alta afinidad para las IgE, pueden activar CDp y transferir ADN a TLR9 en fagosomas. Esta activación da como resultado la secreción de cantidades sustanciales de IFN-α.
Las células T son actores clave en la promoción de la respuesta autoinmune al brindar ayuda a las células B y al activar las células presentadoras de antígenos mediante la liberación de citocinas y el contacto celular directo. Además, se infiltran en los tejidos y promueven la inflamación local. Se supone que las células T CD4 autorreactivas responden a antígenos nucleosomales y, en particular, a péptidos derivados de histonas. Se registró un subconjunto interesante de células T de importancia patogénica desconocida en pacientes con LES y esclerosis múltiple (CXCR3+CD38+CD39+PD-1+HLA-DR+CD161+KLRG1-CD28+OX40+), que difiere de las células TFH y se reconoció por primera vez en el intestino de pacientes con enfermedad celíaca en virtud de la unión al gluten.
Las células TFH promueven la función de las células B y evolucionan a partir de las células T CD4+ en presencia de la IL-6, la IL-21 y el coestimulador inducible de células T (ICOS). La deficiencia de ICOS protege a los ratones MRL-lpr de la enfermedad. Un subconjunto de células CD4+ que se asemeja a las células TFH se expande en la sangre periférica de pacientes con LES activo. El receptor P2X7 ionotrópico controlado por ATP restringe la expansión de las células TFH aberrantes, pero las células TFH de pacientes con LES son resistentes a la inhibición mediada por P2X7 de la expansión impulsada por citocinas, lo que apunta a un defecto de transmisión de señales. Las células T cooperadoras CXCR5-CXCR3+PD-1+, distintas de las células TFH, se encuentran presentes en la periferia y en los tejidos renales de las personas con LES, y ayudan a las células B a producir IL-10 y succinato.
Las respuestas citotóxicas de las células T CD8+ disminuyen en el LES y contribuyen a aumentar las tasas de infección. Una población de células T CD8+CD38+ se expande en la sangre periférica de pacientes con LES. Las células T CD8+CD38+ muestran una producción disminuida de granzimas y perforina y una capacidad citotóxica reducida, y los pacientes con LES, para quienes esta población está expandida, experimentan infecciones con mayor frecuencia. El CD38, un marcador del agotamiento de las células T, es una ectonucleotidasa que degrada el NAD y, por medio de la histona metiltransferasa EZH2, suprime la expresión de moléculas relacionadas con la citotoxicidad. Los inhibidores específicos de la degradación de NAD mediada por CD38 mejoran la disfunción metabólica relacionada con la edad y pueden ser útiles para restaurar la actividad citotóxica de las células T CD8+ en personas con LES. Aunque se argumentó que el agotamiento, definido por los niveles de expresión de las moléculas, es deseable en la autoinmunidad, es importante comprender con mayor detalle los procesos metabólicos implicados.
Las células Treg se caracterizan por la expresión constitutiva del factor de transcripción FoxP3 y expresión alta de la cadena α del receptor de la IL-2 de alta avidez (CD25); en los seres humanos, algunas células efectoras T activadas (Teff) también expresan de manera transitoria esta molécula. El número de células Treg se reduce durante las primeras fases de la enfermedad, mientras que la población de células no Treg CD45RA-FoxP3lo aumenta en el LES activo. La comprensión de que las células Treg tienen receptores de mayor afinidad por la IL-2 y, por lo tanto, una transmisión más fuerte de señales mediada por el receptor de la IL-2 (IL-2R) que las células Teff, sugirió que la administración de IL-2 en una dosis más baja que la utilizada para Teff las células debería promover la expansión y la función de las células Treg. La administración de IL-2 en dosis bajas a ratones propensos al lupus expandió la población de células Treg y redujo el grupo de células T CD3+CD4-CD8- productoras de IL-17, que se sabe que contribuyen al desarrollo de la nefritis lúpica. Se reportó que la IL-2 en dosis bajas administrada a personas con LES produce un beneficio clínico. Una advertencia sobre el éxito aparente de la IL-2 en dosis bajas es la evidencia de que la vía de transmisión de señales IL-2-IL-2R-p-STAT5 en las células T del LES está comprometida. La IL-2 tiene el potencial de revertir varios procesos patogénicos involucrados en el desarrollo del LES, incluida la función deficiente de las células Treg, el aumento de la producción de IL-17, el aumento de la actividad de las células TFH y la expansión de la población de células T CD4-CD8-. La demostración de que las células Treg contribuyen a la reparación de tejidos y la posibilidad de que las células Treg se limiten en los riñones de los pacientes con nefritis lúpica fomenta la consideración de enfoques que enriquezcan las células Treg en los riñones u otros tejidos.
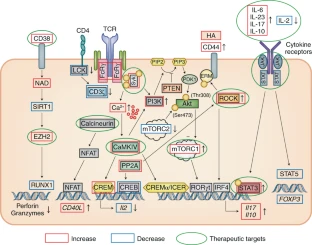
El fenotipo y la función de las células T aisladas de pacientes con LES se estudió de manera amplia en la búsqueda de pistas que expliquen la patogenia de la enfermedad y en un intento de identificar moléculas que puedan servir como biomarcadores y/o dianas terapéuticas. Estos estudios revelaron que, en el contexto del LES, la función de las células T está muy comprometida como resultado de un gran número de aberraciones de transmisión de señales que distorsionan los perfiles de expresión génica y sesgan la respuesta inmune celular hacia un tipo proinflamatorio. En resumen, la transmisión de señales de células T mediada por CD3 es anormal en personas con LES, y esto se sigue por una expresión aberrante de cinasas, fosfatasas, factores de transcripción, receptores de quimiocinas y moléculas de adhesión y la producción de quimiocinas y citocinas proinflamatorias (Fig.2).
Se reconocieron anomalías metabólicas en personas con LES y en ratones propensos al lupus y se relacionó con la función anormal de las células T. Los linfocitos T del LES muestran un mayor estrés oxidativo, como lo indica el agotamiento del glutatión (mediante la pérdida de NADPH), el complejo metabólico de punto de control cinasa mTORC1, la glucólisis y la glutaminólisis. La inhibición del aumento de mTORC1, glucólisis o glutaminólisis mitiga la enfermedad en ratones propensos al lupus.
Nefritis lúpica
El riñón se afecta en más de la mitad de los pacientes con LES y contribuye de manera significativa a la morbilidad. La contribución de los autoanticuerpos con una serie de reactividades y de los complejos inmunes en la expresión de la inflamación renal se revisó de manera amplia a lo largo de los años. Se considera que su papel en la instigación de lesiones está mediado por la activación del sistema del complemento, que explica la respuesta inflamatoria. Los podocitos expresan cantidades aumentadas de la serina/treonina cinasa CaMK4, que, por medio de una serie distinta de eventos bioquímicos, causa daño y la inhibición dirigida a células de CaMK4 en los podocitos evita el depósito de complejos inmunes y nefritis. Estos hallazgos indican la importancia de las células residentes en el inicio y la propagación de la inflamación renal.
La migración de células T al riñón es importante en el desarrollo de una nefritis similar al lupus. Los ratones MRL-lpr que carecen de TCRαβ no desarrollan nefritis lúpica. Aunque se pensaba que las células que infiltran el riñón se encuentran agotadas, las células T CD4+ y CD8+ expandidas de manera clonal con marcadores de células efectoras de memoria se encuentran presentes en los riñones de la nefritis lúpica. Las células T CD8+ se encuentran presentes en todas las muestras de biopsia y se encontró que se adhieren a la cápsula de Bowman e infiltran el epitelio tubular y contribuyen al daño renal. Se encontraron células T productoras de IL-17 en los infiltrados de células renales de pacientes con nefritis lúpica, y la IL-17 es importante para el desarrollo de la nefritis lúpica. Las células TFH se encuentran presentes en los riñones en estrecha asociación con las células B en personas con nefritis lúpica, lo que sugiere que pueden brindarles ayuda. Las células B intrarrenales forman estructuras similares a un centro germinal que produce anticuerpos contra la vimentina, que es un objetivo dominante en la nefritis tubulointersticial en el lupus humano. La aplicación de la metodología de redes neuronales convolucionales profundas en muestras de pacientes con nefritis lúpica permitió el mapeo de la distancia celular, lo que confirmó que las CD presentan el antígeno a las células T CD4+. La comprensión de las arquitecturas celulares de la inmunidad in situ en la nefritis lúpica debería ampliar la comprensión de los procesos patógenos implicados.
Los macrófagos intrarrenales se consideran importantes en el desarrollo de la nefritis lúpica. Los análisis de macrófagos e infiltrados de CD en la nefritis lúpica múrida mostraron una heterogeneidad considerable. Los monocitos se encuentran alrededor de los glomérulos y adyacentes a los túbulos y capilares peritubulares en el intersticio renal y se derivan de la población expandida de monocitos Gr1lo circulantes. La isquemia breve acelera la infiltración de macrófagos inflamatorios Ly6Chi en los riñones de ratones MRL-lpr. Las CD también se infiltran en los riñones en personas con nefritis lúpica, y propaga respuestas inmunitarias adaptativas locales. La infiltración de CD mieloides se asocia con la acumulación de agregados linfoides en los riñones. La identificación de monocitos antiinflamatorios en los riñones de pacientes con nefritis lúpica es importante al considerar enfoques terapéuticos de curación en lugar de inmunosupresores. Por último, los monocitos de patrullaje CD43hiCD11c+F4/80loMHC-II-, que se sabe que orquestan la inflamación renal experimental, se encuentran presentes en los riñones de pacientes con nefritis lúpica y ratones propensos al lupus. Su función depende de la proteína 1 que interactúa con TNFAIP3 (también denominada ABIN1) y su ausencia promueve la nefritis lúpica de una manera dependiente de TLR.
La secuenciación del ARN unicelular de material de biopsia de piel y riñón de pacientes con nefritis lúpica reveló señales de respuesta al IFN tipo I en células tubulares y queratinocitos. Además, la respuesta alta de IFN y las firmas fibróticas en las células tubulares se asociaron con la falta de respuesta al tratamiento. La secuenciación reciente de ARN unicelular de muestras de riñón de personas con nefritis lúpica reveló 21 subconjuntos de leucocitos activos en la enfermedad, incluidas múltiples poblaciones de células mieloides, células T, células naturales asesinas y células B, que demostraron tanto respuestas proinflamatorias como respuestas que resuelven la inflamación. Además, se detectaron evidencias en el riñón de células B activadas y de etapas progresivas de diferenciación de monocitos. Se observó una clara respuesta al IFN tipo I en la mayoría de las células. Dos receptores de quimiocinas, CXCR4 y CX3CR1, se expresaron de manera amplia, lo que implica que pueden tener funciones centrales en el tráfico celular. Estudios similares que investigan las células renales residentes en paralelo pueden revelar cómo la invasión de células inflamatorias altera el panorama de expresión génica y la función de las células renales. La nefritis puede desarrollarse de manera independiente de la autoinmunidad sistémica. Los ratones que carecen de ABIN1 desarrollan glomerulonefritis y autoinmunidad, las cuales dependen de la transmisión de señales de TLR, pero los ratones Rag1-/- y C3-/- deficientes en ABIN1 desarrollan glomerulonefritis sin autoinmunidad. De manera similar, los ratones B6.Nr4a1.Sle1.yaa, que tienen una duplicación del locus Tlr7, carecen de monocitos de patrullaje y son propensos a desarrollar autoinmunidad, no desarrollan glomerulonefritis pero muestran una amplia evidencia de autoinmunidad sistémica. Estudios de cepas NZM2328 propensas al lupus congénito sugirieron antes una falta de asociación entre la autoinmunidad y el daño renal. La noción de que los dos procesos son independientes explica por qué ciertos pacientes con nefritis lúpica desarrollan enfermedad renal en etapa terminal a pesar del tratamiento intensivo con fármacos inmunosupresores, mientras que muchas personas con autoinmunidad sistémica nunca desarrollan enfermedad renal clínica. Existe una multitud de mecanismos que participan en la expresión de la nefritis lúpica, incluidos los complejos inmunes, el complemento, las células T proinflamatorias infiltrantes y las células B productoras de anticuerpos, y los monocitos, y los procesos inherentes de las células residentes explican la mayor parte de la heterogeneidad clínica de la nefritis lúpica y por la respuesta variable a fármacos y biológicos. Comprender el mecanismo operativo dominante en cada paciente es la única forma de desarrollar una medicina personalizada.
LES del sistema nervioso central (SNC)
El sistema nervioso central está involucrado con frecuencia en personas y ratones con LES. Las manifestaciones clínicas son bastante diversas, y reflejan numerosos procesos patógenos inmunes y locales. Hasta ahora, la lesión neuronal mediada por anticuerpos, la activación de células microgliales y la infiltración de células T están involucradas en la expresión de la lesión cerebral. Los anticuerpos que reconocen el ADN de doble hebra pueden reaccionar de forma cruzada con las subunidades NR2A y NR2B del receptor de N-metil-D-aspartato y causar muerte neuronal, de manera principal por medio del aumento de la entrada de calcio neuronal, que imita la excitotoxicidad del glutamato. La transferencia de estos anticuerpos a ratones normales o la inmunización con el pentapéptido DWEYS derivado de NMDAR causa enfermedad neuropsiquiátrica.
Dentro del cerebro, las microglías residentes son las células inmunes predominantes del SNC y son productores potentes de citocinas. Parece que de la lesión neuronal sigue la activación microglial, que implica la activación de la enzima convertidora de angiotensina. El bloqueo de la activación microglial donde un inhibidor de la enzima convertidora de angiotensina limita la lesión neuronal, lo que ofrece otra opción de tratamiento. Los linfocitos y otras células inmunitarias son de manera probable importantes en la expresión de la enfermedad del SNC. Las estructuras linfoides terciarias se encuentran presentes en el plexo coroideo de los ratones propensos al lupus y las personas con LES, y los linfocitos ingresan al parénquima cerebral, pero su naturaleza y función no se estudiaron. Los informes recientes sobre la presencia de células T en el cerebro de personas con autismo y enfermedad de Alzheimer exigen más atención sobre el papel de las células T en la lesión del tejido cerebral.
Lupus cutáneo
Cuatro de los once criterios establecidos por el Colegio Americano de Reumatología para la clasificación de la enfermedad involucran manifestaciones cutáneas que la exposición al sol puede provocar o empeorar. La inflamación de la piel inducida por la luz ultravioleta depende de la producción de la citocina CSF-1 por los queratinocitos, que a su vez recluta y activa a los monocitos, que potencian la apoptosis de los queratinocitos. Los queratinocitos moribundos liberan autoantígenos, incluido Ro60 (los anticuerpos Ro se relacionaron durante mucho tiempo con el lupus cutáneo), que pueden propagar la respuesta autoinmune. Se sabe que los ratones y los seres humanos deficientes en la proteína del complemento C1q tienen un defecto en la eliminación de material apoptótico y tienen manifestaciones cutáneas similares al lupus. Las células apoptóticas recubiertas de C1q son capturadas por las CD, los macrófagos y las células endoteliales al unirse al receptor SCARF1 en la superficie de estas células. Los ratones deficientes en SCARF1 desarrollan una enfermedad similar al lupus.
Las células TH17, que se encuentran presentes en el material de biopsia de piel, pueden contribuir al proceso inflamatorio. Las CDp tienen la capacidad única de producir de manera rápida grandes cantidades de IFN-α tras el reconocimiento de ARN y ADN víricos mediante TLR7 y TLR9 o mediante otros receptores de reconocimiento de patógenos que se encuentran presentes en las lesiones cutáneas. El desarrollo de lesiones cutáneas depende del FasL expresado en la infiltración de células TH1 que reconocen el antígeno afín y de TLR7 en ausencia de TLR9, lo que revela una regulación compleja de la inflamación de la piel en el lupus. Se demostró que las CDp y el IFN-α son abundantes en las lesiones cutáneas.
La importancia del TNF en la expresión de lesiones cutáneas se demostró en experimentos en los que se inyectó IgG de lupus en la piel de varios ratones modificados de manera genética. En resumen, se requerían monocitos, pero no linfocitos o Ig para la inducción de la inflamación de la piel. Aunque se requería TNF, sólo se necesita la trimerización del receptor de TNF tipo I y no la trimerización del tipo II para la inducción de la inflamación. Si bien el bloqueo de la IL-17 puede tener valor en el tratamiento de pacientes con lupus cutáneo, no está claro si los inhibidores de la trimerización del receptor de TNF pueden tener algún valor, de manera particular, dado que el bloqueo de TNF puede conducir a manifestaciones autoinmunes.
Enfermedad cardiovascular en LES
Los pacientes con LES, y en particular aquellos con LDL oxidada y glucoproteína I β2, tienen un riesgo 2 veces mayor de enfermedad cardiovascular. Se encontró que múltiples mecanismos contribuyen a la expresión del daño vascular. Se demostró que el IFN tipo I inhibe la producción de la óxido nítrico sintasa endotelial y causa daño endotelial. Los granulocitos de densidad baja dañan la vasculatura debido a su mayor propensión a NETosis y promueven la fuga vascular y la transición endotelial a mesenquimatosa mediante la degradación de la cadherina endotelial vascular. Las células T efectoras CCR5+T-bet+FoxP3+CD4+ se encuentran presentes en las placas ateroscleróticas. Sin embargo, de manera reciente se afirmó que las células T asesinas naturales invariantes interactúan con los monocitos y promueven un efecto ateroprotector. Mientras que la inflamación promueve la aterosclerosis, se demostró que la hiperlipidemia aterogénica promueve las respuestas de las células TH17 autoinmunes in vivo. Un entorno aterogénico induce la producción de IL-27 por las CD de manera dependiente de TLR4, lo que a su vez desencadena la diferenciación de las células TFH CXCR3+, que se encuentran aumentadas en ratones y pacientes con LES, al tiempo que inhibe la diferenciación de las células T reguladoras foliculares.
Perspectivas y necesidades
Se logró un progreso asombroso en los últimos años debido al aumento de la financiación de diversas fuentes y al reclutamiento de investigadores capacitados de diversas secciones de inmunología y otros campos de la medicina, incluida la nefrología, la dermatología y la cardiología, y otras áreas de la ciencia, incluida la biología molecular avanzada y la bioinformática. Como esta revisión resume, de manera breve, cada aspecto celular, molecular y bioquímico del sistema inmunológico contribuye de alguna manera a la expresión de la enfermedad. En los seres humanos es difícil asignar el tiempo de entrada para cada anomalía registrada en la patogenia de la enfermedad y clasificarla como primaria o secundaria. Los ratones propensos al lupus y los ratones modificados de manera genética, en los que una molécula sospechosa se elimina, sobreexpresa o altera a nivel estructural, demuestran de manera invariable que cada molécula es capaz y suficiente para causar autoinmunidad, lo que refleja un efecto de “castillo de naipes”, en lugar de lo que en realidad sucede en las personas. con el LES. Si bien la comprensión completa de cada vía, ya sea celular o molecular, es de enorme importancia, más importante es la necesidad de dominar la tecnología para identificar qué vía es el impulsor de cada persona con LES.
La lista de ensayos clínicos fallidos sigue en expansión y se actualiza de manera periódica, pero no hubo ningún esfuerzo por cambiar el enfoque de los ensayos clínicos de manera radical. Es lamentable, aunque inevitable, que se defina la enfermedad con diversos criterios de clasificación. No existe duda de que cada biológico ayuda a algunas personas con LES, de manera principal porque esa vía es fundamental para la expresión de la enfermedad en el grupo de pacientes que responden, pero esto no es suficiente. Existen dos soluciones: administrar a cada paciente con LES múltiples fármacos/biológicos con la esperanza de que un mayor número experimente un beneficio clínico, o identificar la vía de conducción en cada paciente y tratarla en consecuencia. Un corolario del primer enfoque es administrar biológicos que corrijan múltiples vías.
El uso cada vez mayor de la secuenciación del exoma completo junto con los estudios de asociación de todo el genoma incrementó el número de pacientes con lupus monogénico, que de manera probable seguirá en aumento. Para estos pacientes, además del tratamiento con medicamentos para controlar las manifestaciones, existe la posibilidad, con el avance de las tecnologías, de hablar de una cura. Aunque todavía es temprano, es importante identificar a los pacientes en los que dos o algunas variantes genéticas contribuyen a la expresión de la enfermedad (pacientes oligogénicos). Los investigadores en el campo deben adaptar las nuevas tecnologías para estudiar cómo los polimorfismos individuales de un solo nucleótido (SNP) interactúan con genes remotos o sus variantes para permitir la patología autoinmune.
El uso cada vez mayor de la secuenciación unicelular ayudará a identificar subconjuntos de células inmunitarias funcionales antes desconocidas que pueden estar en la fase inicial de la desregulación del sistema inmunológico y podrían ofrecer nuevos objetivos para el tratamiento. La secuenciación unicelular de los tejidos involucrados puede revelar más características de las células inmunes invasoras y, lo que es más importante, que las células residentes locales tienen funciones antes no reconocidas y la capacidad de producir moléculas que causan la destrucción de órganos.
Es insondable que, aunque de manera práctica todos los pacientes son mujeres (9 o más de cada 10), se sepa muy poco de lo que se esconde detrás de esto. El cromosoma X contiene numerosos genes involucrados en la respuesta inmune y parece que una inactivación inadecuada puede alterar el equilibrio inmune en favor de la autoinmunidad. ¿Existen reguladores maestros codificados por genes del cromosoma X que comandan todo lo que se reportó sobre el LES? ¿Es necesario que estos genes se expresen de manera homocigótica mediante la liberación del alelo inactivo? En la misma línea de pensamiento, el estudio del epigenoma aún es incipiente en el LES.
Los estudios de los órganos afectados, incluida la piel, el riñón y el cerebro, revelaron procesos locales que son responsables del daño tisular. Sin ignorar el papel de la respuesta autoinmune en la instigación del daño orgánico, es posible que los procesos inflamatorios locales procedan de forma independiente con poca o ninguna información externa. Una mejor comprensión de estos procesos debería abrir nuevos enfoques para el tratamiento de enfermedades.
- Review Article
- Autoimmunity and organ damage in systemic lupus erythematosus
Centro Regional de Alergia e Inmunología Clínica CRAIC, Hospital Universitario “Dr. José Eleuterio González” UANL, Monterrey, México
Dra. Med. Sandra Nora González Díaz Jefe y Profesor
Dr. Carlos Macouzet Sánchez Profesor
Dra. Elma Isela Fuentes Lara Residente 1er Año
Dra. Alejandra Macías Weinmann Profesor




No hay comentarios:
Publicar un comentario
Nota: solo los miembros de este blog pueden publicar comentarios.