Una enfermedad de difícil diagnóstico clínico que suele tratarse con piridostigmina, inmunoterapia y timectomía. Los avances terapéuticos son promisorios.
| Aspectos destacados: |
• El diagnóstico a menudo comienza con pruebas de anticuerpos, mientras que las pruebas de electrodiagnóstico son útiles en pacientes seleccionados. • La piridostigmina se administra a pacientes con síntomas leves o como terapia auxiliar para aquellos con enfermedad más grave. • Los corticosteroides y los agentes ahorradores de corticosteroides se indican en función de una variedad de características del paciente. • La timectomía se reserva principalmente para pacientes más jóvenes con miastenia gravis generalizada con anticuerpos contra el receptor de acetilcolina positivos. |
| Introducción: |
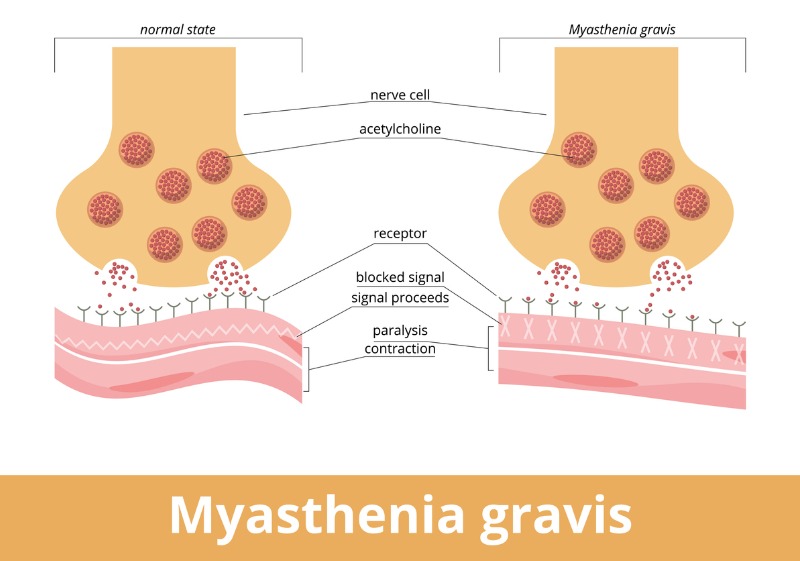
La miastenia gravis es un trastorno neuromuscular autoinmune crónico que causa debilidad del músculo esquelético. Su fisiopatología implica una pérdida de la función del receptor de acetilcolina (AChR) en la unión neuromuscular.
Es más probable que ciertos grupos de músculos esqueléticos estén involucrados que otros, pero el patrón varía ampliamente entre los pacientes y depende del curso clínico individual. En consecuencia, la miastenia gravis generalmente se clasifica como ocular (en la que la debilidad se limita a los músculos oculares extrínsecos y al elevador del párpado superior) o generalizada (en la que están involucrados músculos más allá de los de la forma ocular, incluidos los de las extremidades, el músculo bulbar y la región orofaríngea, y los músculos de la respiración).
Las siguientes 12 preguntas y respuestas frecuentes tienen como objetivo proporcionar información actualizada, de alto rendimiento y clínicamente relevante sobre la miastenia gravis.
1- ¿Qué poblaciones están en riesgo?
Los parientes de primer grado de las personas con miastenia gravis tienen un mayor riesgo no solo de miastenia gravis, sino también de otras enfermedades autoinmunes. La enfermedad tiroidea (tiroiditis de Hashimoto, enfermedad de Graves) es la más frecuente, seguida de la artritis reumatoide.
La enfermedad puede aparecer a cualquier edad, pero el inicio tiene una distribución bimodal, con el primer pico en pacientes en la adolescencia y los 20 años, y el segundo pico en pacientes de 50 y 60 años. La edad de inicio ha aumentado progresivamente, junto con la proporción de hombres, por lo que la preponderancia de mujeres es cada vez menor.
2- ¿Cuándo debe pensar un clínico en este diagnóstico?
Piense en la miastenia gravis cuando un paciente tiene debilidad y fatiga, especialmente de los músculos oculares que produce diplopía variable, ptosis y cierre ocular débil. Estas son las características clínicas centrales.
La debilidad muscular fluctúa, empeorando clásicamente con la actividad física sostenida o repetitiva, al anochecer o durante la noche y mejorando con el reposo. En brazos y piernas, la debilidad suele afectar más a los músculos proximales que a los distales. En la boca y el cuello, se puede observar debilidad bulbar prominente, que incluye disartria, habla nasal, disfagia, control deficiente de la saliva, dificultad para masticar y debilidad del cuello.
Es importante señalar que los pacientes generalmente no tienen síntomas sensoriales o de dolor, disfunción intestinal o vesical, ni cambios en el estado mental o la cognición. Además, los reflejos tendinosos profundos suelen estar intactos. La tabla 1 enumera los trastornos comunes en el diagnóstico diferencial de la miastenia gravis y sus características distintivas.
| Trastorno | Similitudes con miastenia gravis | Diferencias con miastenia gravis |
| Síndrome de Lambert-Eaton | Debilidad y fatiga | Características oculares u oculobulbares menos Prominentes. Arreflexia o hiporreflexia. Características autonómicas (boca seca, disfunción eréctil). Anticuerpo positivo contra el canal de calcio dependiente de voltaje P/Q. La prueba de estimulación nerviosa repetitiva de alta frecuencia muestra una respuesta incremental. |
| Botulismo | Hallazgos oculares (diplopía y ptosis), disfunción bulbar, debilidad generalizada | Ataque agudo, posible antecedente de intoxicación alimentaria. Parálisis descendente. Dilatación de la pupila (midriasis). Disfunción autonómica prominente. Curso monofásico. La prueba de estimulación nerviosa repetitiva de alta frecuencia muestra una respuesta incremental. |
| Esclerosis Lateral amiotrófica | Disfunción bulbar y debilidad | Curso progresivo lento. Sin hallazgos oculares. Los síntomas no fluctúan. Hallazgos de disfunción de la neurona motora superior (p. ej., hiperreflexia, espasticidad). Electromiografía que muestra denervación prominente activa y crónica o reinervación, o ambas. |
| Miopatía | Debilidad de extremidades proximales | Ausencia relativa de hallazgos oculares. Los síntomas no fluctúan. Elevación de creatina quinasa y presencia de anticuerpos específicos de miositis en casos de miositis autoinmune o inflamatoria. La prueba de estimulación nerviosa repetitiva es normal, mientras que la aguja de la electromiografía muestra potenciales de unidad motora polifásicos de corta duración y baja amplitud, con o sin actividad espontánea anormal. |
| Síndrome de Guillain-Barré y polirradiculoneuropatía desmielinizante inflamatoria crónica | Debilidad generalizada | Síntomas sensoriales como dolor y parestesia. Los síntomas no fluctúan. Hiporreflexia o arreflexia El líquido cefalorraquídeo tiene elevación de proteínas, pleocitosis no significativa. Los estudios de conducción nerviosa revelan hallazgos consistentes con desmielinización. |
| Enfermedad ocular de la tiroides | Diplopía | La ptosis es poco frecuente. Los síntomas no fluctúan. Otros hallazgos oculares como edema, enrojecimiento, inyección conjuntival y exoftalmos. Resonancia magnética que muestra ampliación de tejido extraocular. |
| Distrofia muscular oculofaringea | Ptosis, diplopía, disfagia | Curso lentamente progresivo. Ausencia de fluctuación sintomática. Ausencia relativa de debilidad prominente en las extremidades. Elevación de la creatina quinasa. Mutaciones en el gen PABPN1; en su mayoría patrón de herencia autosómico dominante. |
TABLA 1. Características clave que distinguen a la miastenia gravis de otros diagnósticos comunes.
3- ¿Qué pruebas deben solicitarse?
Primero se solicitan pruebas de anticuerpos, seguidas en algunos pacientes por electrodiagnóstico y otras pruebas.
| Pruebas de anticuerpos |
Las pruebas diagnósticas de primera línea suelen ser serológicas.
Anticuerpo anti-AChR es altamente específico (>90%) y muy sensible (hasta alrededor del 85%) en aquellos con miastenia gravis generalizada.
Anticuerpos anti-MuSK. En pacientes con miastenia gravis que son seronegativos para anticuerpos anti-AChR, hasta el 37 % posee anticuerpos anti-MuSK. Sin embargo, la sensibilidad del anticuerpo anti-AChR es más baja, alrededor del 50%, en aquellos que tienen miastenia gravis puramente ocular.
Anticuerpo antiproteína relacionada con lipoproteína 4 (LRP4) se encuentra en el 3% al 50% de los pacientes restantes con miastenia gravis generalizada que son seronegativos a los anticuerpos anti-AChR y anti-MuSK.
Anticuerpos antimúsculo estriado. Son mucho menos específicos para la miastenia gravis y se observan en alrededor del 30% de los pacientes. Son más útiles como marcador de timoma, especialmente en personas que no son de edad avanzada. Por lo tanto, la miastenia gravis no puede ser confiablemente diagnosticada sobre la base de anticuerpo antimúsculo estriado positivo solo.
| Pruebas de electrodiagnóstico |
Dos pruebas de electrodiagnóstico (estimulación nerviosa repetitiva y electromiografía de fibra única) brindan evidencia objetiva del deterioro de la transmisión de la unión neuromuscular y son útiles para diagnosticar la miastenia gravis. No es necesario que se realicen en todos los pacientes, pero brindan evidencia diagnóstica de apoyo, especialmente en aquellos seronegativos y cuando se necesita una rápida confirmación.
Estimulación nerviosa repetitiva utiliza "trenes" repetidos de estimulación nerviosa para generar respuestas musculares eléctricas. La sensibilidad y la especificidad de la estimulación nerviosa repetitiva dependen de las combinaciones de nervio y músculo examinadas, la gravedad de la miastenia gravis y los valores de corte utilizados para una respuesta decreciente. Su sensibilidad diagnóstica general oscila entre un 30 % y un 80 % para la enfermedad generalizada, con menor sensibilidad en la enfermedad más leve o cuando se examinan los músculos distales. En la miastenia gravis ocular, su sensibilidad es solo del 10% al 30%.
Electromiografía de fibra única utiliza pequeños electrodos de aguja para medir la variabilidad de los potenciales de una sola fibra muscular, un reflejo de la transmisión de la unión neuromuscular. Es más sensible que la estimulación nerviosa repetitiva (62% a 99% para la miastenia gravis ocular y 75% a 98% para la miastenia gravis generalizada). Por lo tanto, un resultado normal en un músculo clínicamente débil básicamente descarta la miastenia gravis. Su especificidad informada varía del 66% al 98% para la miastenia gravis ocular y hasta el 98 % para la miastenia gravis generalizada.
| Otras pruebas |
También son útiles en pacientes con sospecha de miastenia gravis las pruebas para enfermedades comórbidas comunes, por ejemplo, tomografía computarizada de tórax o resonancia magnética para anomalías tímicas. Uno debe estar alerta a las características clínicas que pueden sugerir condiciones autoinmunes comórbidas que requerirían pruebas serológicas adicionales, como inmunoglobulina estimulante de la tiroides, peroxidasa antitiroidea, antitiroglobulina o factor reumatoide.
4- ¿Cómo afecta el curso natural al tratamiento?
La miastenia gravis tiende a progresar, especialmente en los primeros años, por lo que recomendamos tratarla de forma agresiva con inmunosupresores al principio y luego ir disminuyendo gradualmente.
Aproximadamente la mitad de los pacientes pueden lograr la remisión o síntomas mínimos con inmunoterapia de dosis baja. Sin embargo, los médicos deben tener cuidado con la interrupción total de la inmunoterapia, ya que solo alrededor del 10% de los pacientes pueden lograr una remisión estable completa sin esta intervención.
5- ¿Qué instrucciones deben recibir los pacientes?
Después de que se diagnostica la enfermedad, se debe informar a los pacientes sobre su curso típico y su pronóstico en gran medida benigno. Los puntos a discutir incluyen:
• Síntomas específicos de la enfermedad, incluidas las señales de alerta.
• La importancia de la tendencia progresiva de la gravedad y frecuencia de los síntomas, en lugar de su empeoramiento transitorio.
• Desencadenantes comunes de la exacerbación, como el calor, la infección, la cirugía, el embarazo, los trastornos emocionales y ciertos medicamentos.
• El régimen de medicación previsto, en particular la inmunoterapia y los posibles efectos secundarios, para garantizar el cumplimiento.
Muchos pacientes con miastenia gravis son cautelosos con el esfuerzo físico, por temor a que el ejercicio empeore sus síntomas. Sin embargo, la mayoría puede tolerar y beneficiarse de alguna forma de actividad. Los pacientes con enfermedad leve pueden participar en entrenamiento de resistencia y aeróbico. Para aquellos con síntomas graves, los ejercicios de estiramiento como el tai chi, el yoga y el entrenamiento del equilibrio suelen ser los más apropiados. El simple hecho de ser más activo y reducir el tiempo sedentario en general es importante.
6- ¿Qué medicamentos es mejor evitar?
Debido a que algunos medicamentos pueden desencadenar o empeorar los síntomas miasténicos, todos los pacientes, especialmente aquellos con debilidad significativa, deben ser observados para detectar un aumento de la debilidad cada vez que se inicia un nuevo medicamento. En principio, si la condición de un paciente se deteriora cuando se le administra un nuevo medicamento, se debe retirar el fármaco.
Drogas que son claramente contraindicadas en la miastenia gravis incluyen telitromicina, magnesio intravenoso, toxina botulínica y penicilamina.
7- ¿Cómo se debe utilizar la piridostigmina?
La piridostigmina, el inhibidor de la acetilcolinesterasa más utilizado para el tratamiento sintomático de la miastenia gravis, suele utilizarse sola en casos leves o en combinación con inmunosupresores en casos más graves. Sin embargo, su eficacia puede ser mínima en pacientes con enfermedad grave o de larga duración.
La dosis de piridostigmina se puede ajustar hasta 240 a 360 mg diarios, pero los efectos secundarios son más comunes en dosis más altas y la sobredosis puede provocar una mayor debilidad. En la práctica, si un paciente necesita más de 240 mg al día, es hora de pasar a la inmunoterapia. Una vez que la miastenia gravis se controla con inmunoterapia, la mayoría de los pacientes no necesitan piridostigmina.
Los efectos secundarios más comunes son gastrointestinales, por ejemplo, calambres abdominales, heces blandas y flatulencia. Bradicardia, broncoespasmo, aumento de la sudoración, lagrimeo excesivo, espasmos musculares y calambres son otros efectos a considerar. Para controlar estas manifestaciones, se pueden tomar glicopirrolato oral o hiosciamina al mismo tiempo que las dosis de piridostigmina.
8- ¿Cuándo deben utilizarse los corticoesteroides?
De acuerdo con las guías de consenso, se deben usar corticosteroides o medicamentos inmunosupresores no esteroideos en todos los pacientes con miastenia gravis que no hayan alcanzado sus objetivos de tratamiento después de una prueba adecuada de piridostigmina.
Los pacientes ambulatorios con síntomas leves a moderados pueden comenzar con 20 mg de prednisona al día y aumentar gradualmente la dosis diaria en 10 mg cada 1 o 2 semanas hasta aproximadamente 60 mg al día, ajustando la dosis según la respuesta clínica. Una vez que se observa una mejoría significativa después de comenzar la terapia con corticosteroides, no hay necesidad de esperar a que ocurra la mejoría máxima antes de comenzar a disminuir estos medicamentos.
9- ¿Cuándo deben utilizarse otros inmunosupresores?
Las terapias inmunosupresoras no esteroides deben considerarse en las siguientes situaciones:
• Falta de respuesta significativa a la prednisona.
• Más de 1 recaída tras la reducción gradual de la prednisona.
• Incapacidad para reducir la prednisona a una dosis mínima aceptable.
• Contraindicaciones de la prednisona, como obesidad mórbida, diabetes mellitus frágil, úlcera péptica, alto riesgo de osteoporosis o efectos secundarios significativos de la prednisona.
Los fármacos inmunosupresores no esteroideos como azatioprina, micofenolato mofetilo, metotrexato, ciclosporina, tacrolimus y rituximab han sido ampliamente utilizado en la miastenia gravis para evitar el uso de corticosteroides. Los agentes más nuevos aprobados recientemente, como eculizumab, ravulizumab y efgartigimod, también podrían cumplir este propósito.
A veces, la terapia inmunosupresora no esteroidea también se puede administrar como inmunosupresor inicial para pacientes con enfermedad leve que están satisfechos con un curso lento de mejoría. En pacientes con debilidad significativa que tienen contraindicaciones para los corticosteroides, la inmunoglobulina intravenosa, el efgartigimod o la plasmaféresis se pueden usar al principio para acelerar la mejoría clínica mientras se deja tiempo para que una terapia inmunosupresora no esteroide alternativa produzca su efecto terapéutico.
10- ¿Cuál es el papel del timo? ¿Quién debe realizarse una timectomía?
La glándula del timo es esencial en el desarrollo de tolerancia central y diferenciación de células T y, por lo tanto, es probable que desempeñe un papel importante en la inmunopatogénesis de la miastenia gravis.
En aproximadamente el 10% de los pacientes, la miastenia gravis es una manifestación paraneoplásica de una neoplasia tímica subyacente (generalmente timoma, rara vez carcinoma tímico). Sin embargo, la hiperplasia linfoide tímica se observa hasta en el 65% de los pacientes con miastenia gravis. La hiperplasia linfoide consta de numerosos linfocitos, macrófagos y células plasmáticas, lo que refleja la autoinmunidad subyacente a la enfermedad grave que a menudo comienza en la glándula del timo.
La timectomía está indicada en todos los pacientes con neoplasias tímicas. De lo contrario, la candidatura para la timectomía depende de varios factores, incluido el estado de los anticuerpos AChR, la severidad, la duración de la enfermedad y la edad del paciente.
Es probable que la timectomía en pacientes adultos similares de 50 años o menos mejore los resultados clínicos y permita una farmacoterapia mínima, incluido el uso y la dosificación de inmunosupresores.
El beneficio de la timectomía en pacientes de 51 a 65 años es más equívoco, y la timectomía generalmente se evita en pacientes mayores de 65 años, ya que la relación riesgo-beneficio es menos favorable.
11- ¿Cómo se puede prevenir, reconocer y tratar la crisis miasténica?
Una crisis miasténica es un empeoramiento potencialmente mortal de la debilidad de los músculos respiratorios o bulbares relacionada con la miastenia gravis que es lo suficientemente severa como para necesitar intubación, ventilación mecánica o ambas.
Las medidas clave para prevenir la crisis miasténica son el control constante de la enfermedad (incluida la adherencia al régimen de medicación y la retirada cuidadosa de los inmunosupresores) y evitar los factores desencadenantes o precipitantes.
| Reconocer la crisis miasténica |
La mayoría de los pacientes con crisis miasténica no se presentan únicamente con insuficiencia respiratoria. Más bien, la debilidad respiratoria neuromuscular por lo general ocurre en el contexto de un empeoramiento de la debilidad generalizada o bulbar. Por lo tanto, las características clínicas que indican un empeoramiento significativo de los déficits en estas áreas pueden proporcionar señales de advertencia.
Es de destacar que las características clásicas de la dificultad respiratoria, como el uso de los músculos accesorios de la respiración, pueden atenuarse durante una crisis, por lo que no se debe confiar demasiado en ellas. La ortopnea es una característica más específica que la disnea, lo que indica una debilidad respiratoria neuromuscular significativa (especialmente del diafragma). La debilidad significativa en los flexores del cuello y los rotadores externos del hombro también se correlaciona típicamente con la debilidad de los músculos respiratorios.
Una prueba de detección que se puede realizar al lado de la cama o por teléfono es la prueba de conteo de una sola respiración. Se le pide al paciente que haga una inspiración profunda y, en la espiración subsiguiente, cuente desde 1 en adelante a un ritmo de habla de rutina (alrededor de 2 cuentas por segundo) hasta que necesiten tomar otro respiro. La incapacidad para contar hasta 20 con una sola respiración indica debilidad respiratoria significativa.
Sin embargo, las medidas espirométricas más formales son ideales y se debe tener en cuenta la “regla 20-30-40”. Esto significa que los pacientes deben ser admitidos o trasladados a la unidad de cuidados intensivos para el manejo respiratorio y de las vías
respiratorias si la capacidad vital cae por debajo de 20 ml/kg, si la presión inspiratoria máxima (también conocida como fuerza inspiratoria negativa) se vuelve menos negativa que -30 cm H2O , o si la presión espiratoria máxima cae por debajo de 40cm H2O.
| Manejo de la crisis miasténica |
El manejo de la crisis miasténica implica optimizar el manejo médico de las enfermedades médicas intercurrentes (incluidas las infecciones), eliminar cualquier medicamento culpable y administrar inmunoterapias agresivas destinadas a mejorar rápidamente la transmisión de la unión neuromuscular.
Las principales terapias son la plasmaféresis (también conocida como recambio plasmático) y la inmunoglobulina intravenosa, pero por lo general no ambas. Tanto la plasmaféresis como la inmunoglobulina intravenosa pueden comenzar a producir mejoras clínicas en varios días. Sin embargo, dado que su eficacia puede comenzar a disminuir en unas pocas semanas, se necesita un aumento concomitante de la inmunoterapia de base (p. ej., corticosteroides). Los medicamentos anticolinesterásicos generalmente se suspenden durante una crisis miasténica, especialmente si el paciente debe ser intubado, ya que la interrupción reducirá las secreciones orofaríngeas y el riesgo de aspiración.
12- ¿Qué tratamientos nuevos hay en el horizonte?
Los inhibidores del complemento eculizumab y ravulizumab y el bloqueador del receptor-Fc neonatal efgartigimod han sido aprobados recientemente por la Administración de Drogas y Alimentos de los EE.UU. para el tratamiento de la miastenia gravis con anticuerpos AChR positivos, y se están estudiando muchos tratamientos más nuevos con diversos mecanismos de acción.
Las inmunoterapias más recientes son generalmente más selectivas en sus objetivos inmunológicos que las más antiguas. En consecuencia, tienen la ventaja de causar menos efectos adversos, incluidas infecciones potencialmente mortales. Sin embargo, son muy caras y un gran inconveniente es su "toxicidad financiera". Para muchos pacientes, las inmunoterapias de amplio espectro más antiguas seguirán siendo un componente clave del tratamiento debido al menor costo, la facilidad de uso y el potencial de inducir la remisión. No obstante, el ritmo de las principales innovaciones terapéuticas en el campo no tiene precedentes y el futuro del tratamiento de la miastenia gravis es prometedor.
Fuente: https://www.intramed.net/




No hay comentarios:
Publicar un comentario
Nota: solo los miembros de este blog pueden publicar comentarios.