
La vasculitis es un grupo heterogéneo de enfermedades caracterizadas por la presencia de inflamación de los vasos sanguíneos. En 1990, el Colegio Americano de Reumatología estableció criterios de diagnóstico para siete vasculitis basados en hallazgos clínicos, biológicos e histológicos, a saber: poliarteritis nodosa (PAN), granulomatosis con poliangeítis (GPA) antes llamada granulomatosis de Wegener, granulomatosis eosinofílica con poliangeítis (GEPA) antes llamada síndrome de Churg-Strauss, angeítis por hipersensibilidad, vasculitis IgA antes llamada púrpura de Henoch-Schönlein, arteritis de células gigantes (ACG) y arteritis de Takayasu. La primera Conferencia Internacional de Consenso de Chapel Hill (CHCC) sobre la Nomenclatura de las Vasculitis Sistémicas en 1994 llegó a un consenso sobre los nombres y definiciones de las formas más comunes de vasculitis, con una actualización en 2012 para mejorar la nomenclatura anterior, cambiar los nombres y definiciones, y añadir categorías de vasculitis que no se incluyeron en la CHCC1994.
Anatomía vascular de la piel
Se requiere un mejor conocimiento de la anatomía vascular de la piel para comprender los diferentes aspectos clínicos de la vasculitis cutánea (Fig. 1). El sistema vascular de la piel consiste en dos plexos interconectados: un plexo profundo y un plexo superficial. Las arterias alimentarias de la piel se sitúan en la hipodermis y se introducen en la dermis profunda donde las arteriolas y las vénulas se comunican para formar el plexo vascular profundo. El plexo profundo se sitúa en la unión dermohipodérmica y también se llama plexo cutáneo. Las ramas de este plexo irrigan el tejido adiposo de la hipodermis, la parte profunda de la dermis y la red capilar que rodea los folículos pilosos, las glándulas sebáceas profundas y las glándulas sudoríparas. Luego, estas ramas pasan de manera vertical a través de la dermis (arterias tipo candelabro). En la dermis superficial, las arteriolas y las vénulas forman un segundo plexo, el plexo superficial o subpapilar, desde el cual una arteria asciende hacia la parte más superficial de la dermis, delante de las papilas dérmicas, y luego desciende por las vénulas hasta llegar al plexo.. Por lo tanto, el plexo subpapilar alimenta la parte superficial de la dermis y la red capilar que rodea los anexos superficiales. Por el contrario, la epidermis no está vascularizada y se alimenta por imbibición de las redes capilares de las papilas dérmicas. Dicha anatomía vascular es importante ya que los infiltrados inflamatorios cutáneos durante la vasculitis se localizan a lo largo de esos vasos, como infiltrados localizados, reagrupados y perivasculares, para diferenciarlos de manera histológica de los infiltrados inflamatorios difusos. Sin embargo, es común que no se visualice la pared de los vasos debido a su destrucción o a los infiltrados.
En lo que respecta a la clasificación, es importante señalar que los vasos medianos según la nomenclatura del CHCC no se presentan en la piel. Sin embargo, en las capas profundas de la piel existen algunos vasos con una pared muscular lisa que se consideran vasos de tamaño medio. No obstante, parece más exacto hablar de vasos pequeños y de vasos con pared muscular lisa en la piel.
Por último, la unidad vascular funcional cutánea es una zona de forma cónica cuya base se encuentra en la superficie de la epidermis y el ápice a nivel de la arteria alimentaria en la unión dermohipodérmica. Esta anatomía vascular explica el aspecto clínico de livedo, donde la obstrucción de la arteria alimentaria conduce a una estasis venosa que dibuja los contornos del cono y que asume un aspecto reticulado.
Detalles de las características histológicas para el análisis de la vasculitis
Tipo de infiltrado inflamatorio
El tipo de infiltrado inflamatorio es un hallazgo clave para el diagnóstico de la vasculitis cutánea.
Vasculitis leucocitoclástica
La vasculitis leucocitoclástica no es una enfermedad en sí misma, sino el resultado de un proceso fisiopatológico común a diferentes causas.. La vasculitis leucocitoclástica refleja un daño relacionado con los complejos inmunes circulantes que se depositan en la pared de los vasos pequeños y activan la cascada del complemento. Las lesiones cutáneas relacionadas con la vasculitis leucocitoclástica se localizan de manera principal en las extremidades inferiores debido a la estasis venosa.
Los diagnósticos histológicos diferenciales de la vasculitis leucocitoclástica son las infecciones y las dermatosis neutrofílicas. El diagnóstico de la dermatosis neutrofílica se basa en los infiltrados dérmicos compuestos de neutrófilos con o sin cuadros de leucocitoclasia. Algunas dermatosis neutrofílicas como el pioderma gangrenoso o el síndrome de Sweet pueden acompañarse de focos de vasculitis como epifenómeno, pero la densidad y la ausencia de angiocentricidad del infiltrado neutrofílico permiten un diagnóstico correcto.
Vasculitis linfocítica
La vasculitis linfocítica puede corresponder a una etapa evolutiva de la vasculitis leucocitoclástica, que marca la transición de una etapa aguda a una subaguda. Sin embargo, algunas vasculitis pueden ser linfocíticas de manera principal, como se observa en la arteritis linfocítica macular. La verdadera vasculitis linfocítica debe diferenciarse de los infiltrados linfocíticos perivasculares sin necrosis fibrinoide de la pared del vaso.
Vasculitis necrotizante
La vasculitis necrotizante se define como la vasculitis acompañada de necrosis fibrinoide. La necrosis tisular se denomina necrosis fibrinoide cuando se asocia con una sustancia extracelular homogénea, eosinofílica, periódica y positiva al ácido-Schiff, debido a la penetración en la pared vascular y en el tejido intersticial de proteínas séricas, como la fibrina, las inmunoglobulinas o el complemento, en especial cuando existen complejos inmunes presentes. La necrosis fibrinoide puede no ser siempre visible, de manera principal debido a la afectación segmentaria o focal como en la PAN o a la delgadez de la pared del vaso afectado en comparación con la intensidad de los infiltrados inflamatorios como en la vasculitis leucocitoclástica.
Vasculopatía trombosante
Durante un proceso inflamatorio se pueden observar varias consecuencias, como la extravasación de glóbulos rojos o las trombosis secundarias debido al daño de las células endoteliales. Sin embargo, la vasculopatía trombosante también puede ser el resultado de un proceso trombótico primario que causa necrosis que induce de manera secundaria a la inflamación. Además, una vasculitis trombosante asociada a un infiltrado mixto o neutrofílico debería sugerir un origen infeccioso, en especial si la leucocitoclasia se encuentra ausente.
Vasculitis granulomatosa
El predominio de los macrófagos con o sin células gigantes caracteriza la vasculitis granulomatosa. El granuloma puede localizarse en la pared del vaso como en la ACG, o ser extravascular como en la GPA. Los diagnósticos diferenciales incluyen trastornos infecciosos y no infecciosos como la sarcoidosis, la metástasis cutánea y la enfermedad de Crohn.
Inmunofluorescencia directa de la piel
La inmunofluorescencia directa de la piel puede revelar inmunoglobulinas (IgA, IgM, IgG), complemento (C3, C1q), o depósitos de fibrinógeno dentro de la pared del vaso. Todas las vasculitis leucocitoclásticas son consecuencia de depósitos de complejos inmunes en la pared de los vasos y, por lo tanto, con inmunofluorescencia directa positiva en la biopsia de la piel. No obstante, esos depósitos evolucionan con el tiempo y la biopsia de una lesión de más de 72 horas suele revelar sólo depósitos de C3 en la pared vascular. Por lo tanto, la inmunofluorescencia directa negativa no elimina la vasculitis leucocitoclástica. La inmunofluorescencia directa puede ser una característica adicional que apoye el diagnóstico de la vasculitis por IgA en caso de depósitos predominantes de IgA en la pared capilar, aunque no es específica. Además, los depósitos de IgM no son específicos y se mencionan de manera convencional en presencia del factor reumatoide o la crioglobulinemia. Por el contrario, los depósitos de inmunoglobulina en la pared vascular sin evidencia de vasculitis no tienen importancia patológica.
Enfoque de diagnóstico según el tamaño de los vasos afectados y las lesiones elementales
Las lesiones cutáneas tienen un gran interés para el diagnóstico de la vasculitis pero no permiten el diagnóstico etiológico. Las lesiones son múltiples y polimórficas, pero pueden proporcionar información valiosa sobre el tamaño de los vasos afectados y el tipo de vasculitis involucrada. De manera aproximada, la mitad de los pacientes con vasculitis cutánea con afectación de los vasos dérmicos o hipodérmicos tienen vasculitis sistémica. Por el contrario, la mitad de los pacientes con vasculitis sistémica desarrollarán manifestaciones cutáneas. La profundidad de los vasos afectados se correlacionó con el tipo de lesiones cutáneas. La afectación de los vasos pequeños superficiales da lugar de manera principal a placas, pápulas y púrpura palpable urticarianas, pero persistentes de manera relativa. La afectación de los vasos de la unión dermohipodérmica o la hipodermis da lugar a úlceras, nódulos o livedo.
Participación de los vasos pequeños
Púrpura
La púrpura es la expresión clínica de la extravasación de glóbulos rojos en la dermis (Fig. 2). La púrpura no siempre es sinónimo de vasculitis relacionada con el daño de la pared del vaso. La púrpura puede ocurrir en ausencia de inflamación de la pared del vaso. Una púrpura no palpable de manera inicial, oscura, con evolución necrótica, se traduce más a menudo en vasculopatía trombótica que en vasculitis. Por el contrario, una púrpura palpable suele reflejar la presencia de un infiltrado de células inflamatorias. La púrpura papulomatosa o polimórfica suele ser la expresión clínica de la vasculitis leucocitoclástica. Sin embargo, algunas vasculitis pueden resultar en púrpura no palpable..
Pápulas no purpúricas
Dentro de la vasculitis, las pápulas suelen reflejar la afectación de los vasos de tamaño pequeño de la dermis. Las pápulas corresponden a lesiones palpables y sólidas que miden menos de 1 cm. La lesión de vasculitis se hace palpable debido a la intensidad del infiltrado inflamatorio, por lo general dentro de las áreas perivasculares superficiales y/o profundas. Estas lesiones son casi siempre eritematosas debido a la vasodilatación causada por el infiltrado perivascular. Además, el edema asociado de la dermis puede ser responsable por sí mismo de la lesión palpable, como en la urticaria, o coexistir con una infiltración celular. Las pápulas también pueden reflejar lesiones granulomatosas asociadas a la vasculitis, en especial durante la GPA o la GEPA. En estas situaciones, las pápulas son eritematosas o moradas, localizadas en las áreas de extensión del codo y los dedos, y por lo general son múltiples y simétricas.. Estas lesiones pueden parecerse a vesículas, nódulos o ulceraciones. De manera histológica, existe una necrosis endotelial con un infiltrado granulomatoso extravascular compuesto por eosinófilos, linfocitos e histiocitos y la necrobiosis de los haces de colágeno suele ser basofílica en la GPA y eosinofílica en la GEPA.
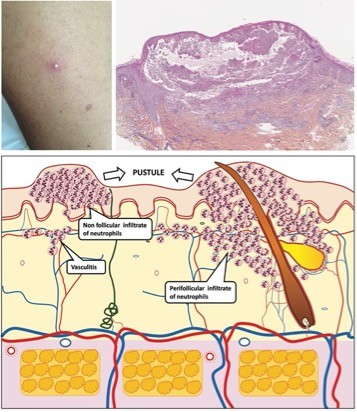
Pústulas
Las pústulas son causadas por los desprendimientos de la epidermis cuyo contenido se constituye por neutrófilos. Las pústulas son a veces secundarias a la púrpura y a la intensidad del infiltrado inflamatorio. Las pústulas foliculares o no foliculares con contorno púrpura reflejan la vasculitis de los pequeños vasos y se observan de manera típica en la enfermedad de Behcet o en las enfermedades inflamatorias del intestino. También pueden aparecer durante una endocarditis infecciosa o una infección gonocócica.
Participación de los vasos de la pared del músculo liso (vasos de tamaño medio)
Nódulos
Los nódulos corresponden a lesiones palpables y sólidas que miden más de 1 cm. Por lo general, los nódulos se localizan en la dermis o la hipodermis. En el contexto de la vasculitis, reflejan la afectación de los vasos de la unión dermohipodérmica o de la hipodermis (Datos complementarios Fig. 2). Los nódulos pueden situarse a lo largo de una arteria o una vena superficial, y pueden evolucionar hacia una úlcera o necrosis.
Para una evaluación patológica adecuada de un nódulo se requiere una biopsia profunda con el bisturí, que retire la hipodermis. La vasculitis asociada a los nódulos cutáneos afecta a los vasos con una pared muscular lisa como en la PAN, o la vasculitis que afecta a los vasos capilares y a los vasos de la pared muscular lisa como la vasculitis asociada a ANCA.
Livedo
El livedo corresponde a un eritema reticulado (en forma de red) de origen vascular. El livedo es secundario a los trastornos vasomotores o a la patología vascular obstructiva, con o sin afectación parietal. No es sinónimo de vasculitis. De manera clínica, existen diferentes tipos de livedo:
- Livedo reticularis, con mallas finas, regulares y cerradas, fisiológico de manera frecuente;
- Livedo racemoso, con mallas rotas, ramificadas, discontinuas e irregulares, siempre patológicas y características del síndrome de Sneddon;
- Livedo con mallas infiltradas, de manera frecuente encontrado durante la vasculitis (PAN);
- Livedo purpúrico o púrpura retiforme que se dirige hacia una patología trombosante de los
vasos dérmicos como una coagulación intravascular diseminada o el síndrome antifosfolípidos catastrófico.
En general, el livedo reticularis es fisiológico más a menudo, mientras que un livedo racemoso es siempre patológico y requiere una evaluación minuciosa. La biopsia de piel no es sistemática y su interés es debatido. No existe interés de la biopsia de piel en el livedo reticularis, mientras que en el livedo racemoso con infiltración o necrosis, la biopsia de la zona infiltrada a veces puede revelar vasculitis. En presencia de un livedo racemoso no infiltrado y no necrótico, limitado a las extremidades inferiores, la biopsia no suele ser contributiva, pero a veces puede mostrar un cuadro de endarteritis obliterante que apunta a un diagnóstico de síndrome de Sneddon, o un aspecto fibrinoide hialino intenso de la pared de una arteria muscular observado en la arteritis linfocítica macular.
Manifestaciones isquémicas: gangrena o necrosis de las extremidades, la piel cabelluda o la lengua
La necrosis de la piel es el resultado de la oclusión de los vasos sanguíneos de la piel. La extensión de la necrosis depende del tipo y la profundidad de los vasos afectados. Las lesiones necróticas se observan con frecuencia en la crioglobulinemia tipo I, donde el principal mecanismo es una vasculopatía trombótica. Tales lesiones también pueden observarse durante la vasculitis asociada a ANCA y la arteritis digital necrotizante durante la PAN. La necrosis del cuero cabelludo y/o la lengua se produce en la ACG.
Otras manifestaciones asociadas con vasculitis
Pioderma gangrenoso
El pioderma gangrenoso es una ulceración cutánea dolorosa, localizada de manera predominante en los miembros inferiores, con extensión centrífuga progresiva que puede alcanzar más de 10 cm, y que comienza de manera inicial como una pústula o nódulo subcutáneo de novo o después de un traumatismo leve, con bordes elevados inflamatorios y violáceos y algunas zonas con conejeras purulentas. El pioderma gangrenoso suele presentarse en pacientes con enfermedad inflamatoria intestinal, pero también puede presentarse en pacientes con GPA y arteritis de Takayasu.
Tromboflebitis superficial
La tromboflebitis superficial debe distinguirse de la tromboflebitis profunda y de la trombosis de várices. El cuadro clínico incluye nódulos sensibles, a veces como una lesión lineal profunda o un cordón mal limitado. Tales lesiones pueden observarse en trastornos trombofílicos como el síndrome de antifosfolípidos o la deficiencia de proteína C o S, o en la vasculitis, principalmente la enfermedad de Behçet. En otros lugares, pueden ser el modo revelador de los cánceres sólidos, en especial en el estómago o el páncreas.
Hipertrofia gingival
La hipertrofia gingival, con aspecto de frambuesa, se describió como una manifestación mucosa de la GPA, que es la única vasculitis asociada a esta característica.
Úlceras orales, genitales o anales
Las úlceras orales son manifestaciones poco frecuentes de la vasculitis, excepto en el caso de la enfermedad de Behçet, en la que representa la manifestación más común. Las úlceras orales también pueden observarse de manera rara en la GPA (5% de los pacientes) o en la policondritis recidivante, de manera especial dentro de la boca y las úlceras genitales con síndrome de cartílago inflamado (MAGIC).
Hemorragias en astilla
Las hemorragias en forma de astilla, pequeñas áreas de hemorragias debajo de las uñas de las manos o de los pies, son raras en la vasculitis y es más probable que se deban a una patología tromboembólica o trombótica, de manera especial durante el síndrome antifosfolípidos catastrófico o la endocarditis infecciosa.
Fenómeno de Raynaud
El fenómeno de Raynaud se observa de manera principal en las enfermedades del tejido conectivo, principalmente en la esclerodermia sistémica, la enfermedad mixta del tejido conectivo y la miopatía inflamatoria. En la vasculitis, el fenómeno de Raynaud se dirige hacia la vasculitis crioglobulinémica, en hasta 30% de los pacientes con crioglobulinemia tipo I y de manera aproximada 20% en la vasculitis crioglobulinémica mixta.
Dermatitis granulomatosa intersticial
La dermatitis granulomatosa intersticial puede presentarse de manera clínica como cordones lineales de las regiones axilares, pero también pápulas y placas. De manera histológica, se observan de forma típica focos de necrobiosis dérmica profunda, que afectan a uno o unos pocos haces de colágeno y se rodean de histiocitos organizados en rosetas. Esta entidad se relaciona de manera nosológica con los llamados granulomas de Churg-Strauss y se asocia con muchos trastornos, como la GEPA y MPA, la artritis reumatoide, el lupus eritematoso sistémico y la enfermedad de Still de inicio en la edad adulta.
Descripción de las principales vasculitis cutáneas
Vasculitis de vasos pequeños de la piel
Angeítis leucocitoclástica cutánea
La vasculitis por hipersensibilidad o angeítis leucocitoclástica cutánea se individualizó en el CHCC1994 y el CHCC2012 y pertenece a la vasculitis de un solo órgano. De manera clínica, las manifestaciones cutáneas de la angeítis leucocitoclástica cutánea son polimórficas e incluyen púrpura, placas urticariales, nódulos, vesículas o bullas, pústulas y/o ulceración.
Vasculitis urticariana
La vasculitis urticariana se define de manera histológica por la inflamación de los capilares de la dermis y los vénulas poscapilares, y se divide en dos grupos según el nivel de fracción del complemento: vasculitis urticariana normocomplementémica y vasculitis urticariana hipocomplementémica.. Las lesiones de la vasculitis urticariana se diferencian de la urticaria clásica porque son más fijas, son menos pruriginosas, y persisten por más de 24 h. Tienen una evolución crónica, que deja máculas hiperpigmentadas, y se asocian a angioedema en la mitad de los casos, púrpura en un tercio, y livedo en 14%. Pueden presentarse manifestaciones extracutáneas, de manera principal artralgias, y afectación ocular, pulmonar, gastrointestinal y/o renal. La mayoría de las vasculitis urticarianas normocomplementémicas son idiopáticas, mientras que la vasculitis urticariana hipocomplementémica se asocia de manera frecuente con enfermedades del tejido conectivo. La vasculitis urticariana debe distinguirse de la dermatosis urticariana neutrofílica, ya que muchos casos de UV se reclasificaron como dermatosis neutrofílica urticarial durante la última década.
Vasculitis IgA (púrpura de Henoch-Schönlein)
La vasculitis por IgA se caracteriza por púrpura palpable, artralgia y dolor abdominal, y representa 10% de las vasculitis cutáneas. La vasculitis por IgA es la vasculitis más frecuente en la infancia, con una incidencia anual de 3 a 26 por cada 100,000 niños, y suele ocurrir entre los 4 y los 7 años. La enfermedad es menos frecuente en los adultos, con una incidencia estimada entre 0.1 y 1.8 por cada 100,000 personas. La manifestación dermatológica tanto de la vasculitis leucocitoclástica como de la vasculitis por IgA es la púrpura palpable. Michel y colaboradores describieron los criterios clínicos para diferenciar la vasculitis IgA de la angeítis leucocitoclástica cutánea. La presencia de 3 de los siguientes 6 criterios clasifica de manera correcta a un paciente como poseedor de vasculitis IgA en 87%: púrpura palpable no relacionada con la trombocitopenia, dolor abdominal difuso posprandial de manera típica asociado con diarrea sanguinolenta, hemorragia gastrointestinal, hematuria, edad inferior a 20 años y ausencia de fármacos al comienzo de la enfermedad que pueden precipitar la afección.
De manera histológica, la vasculitis IgA no muestra ninguna especificidad, que muestra la típica vasculitis leucocitoclástica. Sin embargo, la inmunofluorescencia directa revela depósitos predominantes de IgA en la pared capilar. Por lo tanto, el predominio de los depósitos perivasculares de IgA en la biopsia de piel es un argumento positivo adicional para el diagnóstico de la vasculitis IgA, sin ser específico, ya que otras vasculitis como la vasculitis crioglobulinémica pueden tener depósitos de IgA.
Vasculitis crioglobulinémica
La crioglobulinemia se define por la presencia de inmunoglobulinas que se precipitan con las temperaturas frías y se resuelven con el calentamiento. La clasificación se basa en el análisis inmunoquímico, que define tres tipos. Las crioglobulinas tipo I son inmunoglobulinas monoclonales únicas siempre vinculadas a un trastorno linfoproliferativo de células B. Las crioglobulinas tipo II consisten en IgG policlonal con IgM monoclonal con actividad de factor reumatoide. Las crioglobulinas tipo III se componen por IgG e IgM policlonales con actividad de factor reumatoide. Los tipos II y III se suelen denominar crioglobulinemia mixta (CM) y pueden relacionarse con el trastorno linfoproliferativo de células B, trastornos autoinmunes y/o infecciones. La crioglobulinemia mixta es responsable de la vasculitis con depósitos inmunes de crioglobulina que afectan a los vasos pequeños (capilares, vénulas o arteriolas). La púrpura vascular es la manifestación más frecuente, a menudo indicativa de la enfermedad, mientras que la neuropatía periférica se presenta en la mitad de los pacientes y la afectación renal en un tercio de ellos. En cambio, la crioglobulinemia monoclonal tipo I es responsable de la oclusión vascular con frecuentes fenómenos de Raynaud, úlceras y necrosis distal, dolor e hinchazón en las extremidades o síndrome de hiperviscosidad. De manera histológica, la crioglobulinemia se presenta sobre todo con un infiltrado de células mononucleares perivasculares, sin que la pared del vaso se afecte por sí misma. Sin embargo, es posible que se produzca una vasculitis leucocitoclástica, en especial dentro de la piel, con necrosis fibrinoide de la pared de los vasos de pequeño tamaño e infiltrado inflamatorio de neutrófilos, algunos de ellos picnóticos (leucocitoclasia). Se observan depósitos hialinos intravasculares, con presencia de inmunoglobulina y depósito de complemento mediante inmunofluorescencia directa.
Vasculitis de vasos pequeños de la piel y de los vasos de la pared del músculo liso
La asociación de las lesiones vasculíticas de vaso pequeño y de vasos con pared de músculo liso es muy sugerente de la vasculitis asociada a ANCA, es decir, GPA, MPA y EGPA.
Granulomatosis con poliangeítis
La granulomatosis con poliangeítis se caracteriza de manera histológica por la inflamación de la pared del vaso con granulomas peri o extravasculares.. De manera clínica, la GPA se caracteriza por manifestaciones en oído, nariz y garganta, afectación pulmonar y glomerulonefritis. Los ANCA son positivos en más de 90% de los casos, dirigidos contra la proteinasa 3. Sí, el hallazgo cutáneo más común es la púrpura palpable relacionada con vasculitis leucocitoclástica.
Poliangeítis microscópica
La poliangeítis microscópica es responsable de una glomerulonefritis necrosante segmentaria y focal, asociada a proliferación extracapilar, y no a un granuloma extravascular. Otras manifestaciones clínicas incluyen artralgias, neuropatía periférica, hemorragia alveolar o manifestaciones gastrointestinales. Los ANCA son positivos en más de 80% de los casos, dirigidos contra la mieloperoxidasa.
Granulomatosis eosinofílica con poliangeítis
La granulomatosis eosinofílica con poliangeítis es una vasculitis necrosante definida por la asociación de asma, eosinofilia sanguínea y tisular y vasculitis sistémica. Las manifestaciones clínicas incluyen mononeuropatía o polineuropatía múltiple, infiltraciones pulmonares no fijas, anomalías sinonasales y cardiomiopatía. Las manifestaciones cutáneas son frecuentes y se dan hasta en 50% de los pacientes, en especial la púrpura, los nódulos subcutáneos y la urticaria. Se notificaron manifestaciones menos frecuentes, como la celulitis de Wells, la enfermedad de Kimura y el granuloma de Lever, todas ellas pertenecientes al espectro de las dermatosis eosinofílicas.. De manera histológica, el fenómeno de Wells puede estar presente, ya que las figuras eosinofílicas en llama corresponden a la destrucción del colágeno por degranulación de los eosinófilos. Los ANCA son positivos en sólo 30-40%, de manera habitual dirigidos contra la mieloperoxidasa.
Vasculitis cutánea de los vasos de la pared de músculo liso
Poliarteritis nodosa
La poliarteritis nodosa es una vasculitis necrosante segmentaria y focal que afecta a las arterias medianas y pequeñas y a las arteriolas de la dermis profunda y los septos, con un infiltrado inflamatorio neutrofílico responsable de manera frecuente de la trombosis vascular. La enfermedad puede ser sistémica con afectación cutánea (PAN sistémica), y limitarse a la piel como vasculitis de un solo órgano (PAN cutánea); en esta última, sin embargo, se produce con frecuencia una afectación neurológica regional.
Las manifestaciones cutáneas de la PAN sistémica se describen en casi 50% de los casos, en los que predominan la púrpura (19%), el livedo (17%), los nódulos (15%), la urticaria (6%) y las úlceras o la necrosis cutánea (4%).. Aunque la púrpura se menciona a menudo en los estudios clínicos, es incompatible con la definición actual de la CHCC2012, ya que es una manifestación de la vasculitis de vasos pequeños, el tipo de vasos que se salvan en la PAN. Las posibles explicaciones serían que estas PAN con púrpura serían MPA o vasculitis crioglobulinémica, o por el contrario, una definición inexacta de PAN en el CHCC2012.
Durante la PAN cutánea, las manifestaciones de la piel son de manera principal livedo, nódulos y úlceras que afectan de forma predominante a las extremidades inferiores. Se describieron otras manifestaciones, como atrofia blanca, fenómeno de Raynaud y placas inflamatorias rodeadas de nódulos. Se identificaron factores desencadenantes de la PAN cutánea, como la infección viral, bacteriana o micobacteriana, la enfermedad inflamatoria intestinal o los medicamentos (minociclina).
En 2003 se describió otra entidad, denominada arteritis linfocítica macular o arteritis trombofílica linfocítica (ALM). Los autores informaron de manera inicial de la asociación de una dermatosis macular y pigmentada con la arteritis linfocítica en la hipodermis o la dermis profunda en la biopsia de piel. De manera clínica, las lesiones cutáneas son máculas eritematosas y/o pigmentadas, reticuladas, a veces asociadas a nódulos y púrpura. Las lesiones siempre se localizan en los miembros inferiores y con menor frecuencia en los miembros superiores (44%) y de manera más rara en el tronco. La ALM se presenta de manera preferente en mujeres en 70-80% y la edad media es de 40 años. De manera histológica, la epidermis y la dermis son normales; se observa un denso infiltrado linfocítico peri e intravascular que rodea las arteriolas pequeñas en la dermis reticular o hipodermis. No se observa destrucción ni necrosis en la pared arterial, pero un anillo arterial circunferencial fibrinoso e hialino muy homogéneo es característico de la ALM. Se pueden encontrar autoanticuerpos, de manera principal anticardiolipina y anticuerpos antinucleares, a veces con especificidad anti-SSA. Sin embargo, esta entidad es controvertida y no se estableció una distinción real entre PAN y ALM cutáneas. En 2015, Buffiere-Morgada y colaboradores evaluaron de manera retrospectiva y a ciegas, la frecuencia de las características clínicas e histológicas de la ALM en los pacientes a los que se les diagnosticó PAN cutánea. La arteritis linfocítica predominante, la escasez de neutrófilos, el anillo de fibrina concéntrico y la ausencia de disrupción elástica de la lámina interna estuvieron presentes en 60%, 20%, 18% y 23% de los pacientes, de manera respectiva. La incidencia de remisión completa no es diferente entre los pacientes con un infiltrado predominante de linfocitos o con pocos neutrófilos. Estos datos no favorecieron la clasificación de la PAN y la ALM cutánea como entidades distintas..
Enfermedad de Kawasaki
La enfermedad de Kawasaki es una vasculitis de vasos grandes y medianos, de etiología desconocida, que afecta de manera preferente a niños de menos de 5 años, y de manera más rara a adultos. La enfermedad de Kawasaki resulta en un síndrome adenocutáneo-mucoso febril. Su gravedad se relaciona con la afectación cardíaca y el desarrollo de aneurismas coronarios.
Enfermedad de Behçet
La enfermedad de Behçet es una vasculitis inflamatoria multisistémica que afecta a las arterias y venas de tamaño variable, y de manera preferente a los hombres jóvenes de unos 30 años. Además de las frecuentes úlceras bucales y genitales, las manifestaciones cutáneas incluyen nódulos inflamatorios localizados en las extremidades inferiores y que se asemejan al eritema nodoso, “seudofoliculitis” y tromboflebitis superficial. Los ataques oculares, gastrointestinales, vasculares o del sistema nervioso central son menos frecuentes, pero mucho más graves.
Vasculitis inducida por medicamentos con depósito de complejos inmunes
La vasculitis inducida por medicamentos con depósito de complejos inmunes se individualizó en el CHCC2012 como una vasculitis de causa definida. Ocurre más a menudo dentro de los 7 a 21 días después del inicio del medicamento sospechoso. Los medicamentos involucrados de manera más frecuente son la fosfenitoína, la quinidina, las sulfonamidas, las penicilinas, el alopurinol o el factor estimulante de colonias de granulocitos. En general, de manera aproximada, 15-20% de la vasculitis cutánea es inducida por medicamentos.
Cómo se maneja a un paciente con sospecha de vasculitis cutánea
Cuando se sospecha la existencia de una vasculitis cutánea, es obligatorio realizar un diagnóstico cuidadoso para buscar manifestaciones orgánicas graves de manera potencial y establecer lo antes posible el diagnóstico etiológico:
1. Realizar una biopsia de piel de una lesión temprana con histología estándar e inmunofluorescencia directa
2. Determinar la gravedad de la vasculitis al buscar manifestaciones sistémicas: síntomas constitucionales, artralgias, mialgias, manifestaciones pulmonares, síntomas gastrointestinales, manifestaciones en oídos, nariz y garganta, signos oculares, neuropatía periférica, afectación renal y manifestaciones urogenitales (análisis de orina anormal, dolor testicular)
3. Identificar una causa:
a) ¿Iniciación reciente de los medicamentos?
b) ¿Una infección reciente?
c) ¿Neoplasia, hemopatía maligna?
d) Pruebas de laboratorio: CBC, plaquetas, CRP, PT, APTT, fibrinógeno, electrolitos séricos, urea, creatinina, pruebas de función hepática, electroforesis de proteínas séricas, serologías de VIH, VHB y VHC, serología de parvovirus B19 (y/u otros virus según el entorno clínico y epidemiológico), crioglobulina, factor reumatoide, niveles de fracciones de complemento, anticuerpos antinucleares y, en caso de ser positivo, pruebas de ANCA, proteinuria y análisis de orina
e) Investigaciones morfológicas: radiografía de tórax
f) Investigaciones que se debatirán en función del contexto clínico y la evolución de la enfermedad: hemocultivos, ecocardiografía, tomografía computarizada y sinusitis, imágenes del cerebro y punción lumbar.
Conclusión
La vasculitis cutánea constituye un grupo grande y heterogéneo de enfermedades, para el cual el análisis de las lesiones elementales cutáneas y la biopsia de piel proporcionan importantes hallazgos sobre el tamaño del vaso afectado y de manera posible sobre el tipo de vasculitis.
Centro Regional de Alergia e Inmunología Clínica CRAIC, Hospital Universitario “Dr. José Eleuterio González” UANL, Monterrey, México
Dra. Med. Sandra Nora González Díaz Jefe y Profesor
Dra. Hilda Hernández Sánchez Profesor
Dra. Alejandra Canel Paredes Residente 1er Año
Dra. Alejandra Macías Weinmann Profesor




No hay comentarios:
Publicar un comentario
Nota: solo los miembros de este blog pueden publicar comentarios.