INTRODUCCIÓN
La vasculitis de vasos pequeños de la
piel, que se presenta de forma clásica y más común como una púrpura palpable en
las extremidades inferiores, se denomina de manera intercambiable con un número
de términos, cada uno de los cuales lleva un tono ligeramente diferente del
significado. Estos incluyen “vasculitis cutánea leucocitoclástica”, o simplemente
“vasculitis leucocitoclástica”, “vasculitis por hipersensibilidad”, “vasculitis
leucocitoclástica cutánea” y “vasculitis de vasos pequeños de la piel”, el
término para la vasculitis limitada a los vasos pequeños de la piel aceptado y revisado
de manera reciente por los Criterios del Consenso Chapel Hill en 2012.
De forma independiente de la terminología
utilizada, es importante tener en cuenta que la presentación clínica de la
vasculitis de vasos pequeños de la piel debe considerarse inicialmente un síntoma
más que una entidad por sí misma. En otras palabras, cuando se diagnostica
vasculitis limitada a la piel, primero hay que descartar posibles
manifestaciones sistémicas (como el involucro de articulaciones, renal o
gastrointestinal), causas subyacentes, y las asociaciones de enfermedades que
afectan al tratamiento y el pronóstico. Además, los pacientes pueden comenzar
con una enfermedad limitada a la piel y desarrollar manifestaciones sistémicas
con el tiempo, lo que exige un seguimiento cuidadoso. Un subconjunto específico
de vasculitis de vasos pequeños de la piel merece una mención especial; la
vasculitis por inmunoglobulina IgA (también conocida como la púrpura de
Henoch-Schönlein). Es un síndrome mediado por IgA caracterizado por el involucro
cutáneo, gastrointestinal, articular, y/o afectación renal. Aunque la
presentación inicial de esta condición puede ser indistinguible de la
vasculitis de vasos pequeños de la piel no mediada por IgA, su tratamiento y
pronóstico es diferente. En general, sin embargo, la vasculitis de vasos
pequeños de la piel es más a menudo aguda y autolimitada, y su pronóstico es
favorable, sobre todo cuando está ausente la participación interna.
EPIDEMIOLOGÍA
La vasculitis de vasos pequeños de la
piel afecta a ambos sexos por igual y pacientes de todas las edades. Estudios
realizados en España reportan una incidencia anual de 30 casos de vasculitis
por hipersensibilidad por millón de adultos por año. Por el contrario, la
vasculitis por IgA tiene una incidencia de 14 casos por millón de adultos por
año. Un estudio reciente, basado en la población en Minnesota encontró que la
incidencia de vasculitis cutánea leucocitoclástica (incluyendo vasculitis por
IgA, así como otros tipos de vasculitis de vasos pequeños) era casi idéntica,
en 45 casos por millón. En los niños, por el contrario, la vasculitis por IgA es
mucho más común que la no mediada por IgA. La presencia de una vasculitis
sistémica asociada subyacente, enfermedad del tejido conectivo, o malignidad es
mucho más común en adultos que en niños.
FISIOPATOLOGÍA
La vasculitis de vasos pequeños de la
piel está mediada por el depósito de complejos inmunes en los vasos afectados. Antígenos
circulantes debidos a medicamentos, infecciones, enfermedades del tejido
conectivo, o neoplasias se unen a anticuerpos, y hay formación de complejos inmunes
que se alojan y se atrapan dentro de los vasos pequeños, ya sea en la dermis
superficial, con mayor frecuencia en las zonas de declive, las articulaciones,
el tracto gastrointestinal, o los glomérulos. Estos complejos, a su vez,
activan el complemento e inducen una respuesta inflamatoria que conduce a la
destrucción de los vasos y la extravasación de las células rojas de la sangre.
En el caso de la púrpura palpable en la piel, este involucro de los vasos
pequeños explica el tamaño pequeño (usualmente) de las lesiones; la cascada del
complemento y la inflamación subsiguiente explican que las lesiones sean
palpables y la sintomatología (que a menudo arden); y la extravasación de
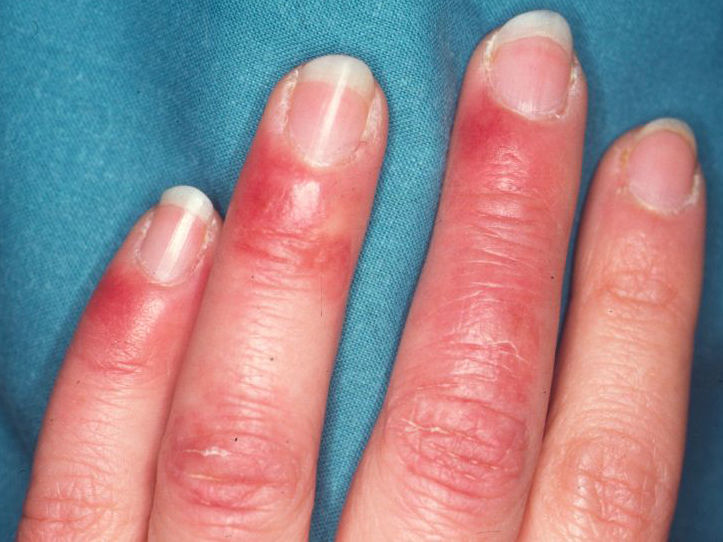
células rojas resulta en la púrpura no escaldada (Fig. 1).
ETIOLOGÍA
Alrededor de la mitad de los casos son
idiopáticos. El resto son más a menudo inducidos por fármacos o postinfecciosos.
Los antibióticos, y los b-lactámicos en particular, son causas comunes, pero casi
cualquier medicamento o aditivo de medicamentos puede causar vasculitis. Entre
las causas infecciosas, las infecciones respiratorias superiores (como el Streptococcus β-hemolítico del grupo A)
y la hepatitis C se implican de forma común; sin embargo, se describen numerosos
factores infecciosos desencadenantes. Determinar una causa específica puede ser
difícil, sobre todo en el entorno hospitalario, cuando muchos pacientes tienen
tanto una historia de infección reciente y la exposición a numerosos
medicamentos.
Aunque la púrpura palpable es más a
menudo debida a una infección o un medicamento, es importante recordar que la vasculitis
de vasos pequeños de la piel también puede ser debida a una enfermedad subyacente
del tejido conectivo, como el lupus eritematoso sistémico, elsíndrome de Sjögren,
la artritis reumatoide o la dermatomiositis, y puede, de hecho, ser el signo de
presentación de dicha enfermedad. La vasculitis debida a enfermedad subyacente del
tejido conectivo puede asociarse con manifestaciones internas más
significativas. Las manifestaciones cutáneas de la vasculitis de vasos pequeños
como la púrpura palpable y lesiones de urticaria también pueden ser una
característica de la vasculitis asociada a anticuerpos anticitoplasma de
neutrófilos (ANCA) con afectación sobrepuesta de vasos de pequeño y mediano
calibre de la piel. Un pequeño porcentaje de los pacientes (<5%) puede tener
un malignidad hematológica subyacente o de órgano sólido. La vasculitis tiende
a aparecer 7 a 10 días después de la exposición a un fármaco o un
desencadenante infeccioso y con una media de 6 meses después de la aparición de
una condición médica subyacente. En la práctica, sin embargo, el alcance y el
momento de aparición varían en gran manera.
La infección respiratoria viral o la
faringitis estreptocócica con frecuencia preceden a la aparición de la vasculitis
por 1 a 2 semanas. En general, alrededor de 40% de los casos son atribuibles a
una causa infecciosa, de los cuales muchos se reportan. La exposición a
medicamentos puede ser el culpable en alrededor de 20%. En los adultos, puede
considerarse la IgA paraneoplásica; 90% de estos pacientes son masculinos. Con
la vasculitis de vasos pequeños de la piel como un todo, una fracción
significativa de los casos no tiene causa identificable.
Los pacientes con vasculitis cutánea que
no es autolimitada o que es refractaria o recurrente pueden ser más propensos a
tener una enfermedad médica subyacente que conduce a la vasculitis. En tales
casos, el abordaje adicional puede ser necesario para dilucidar la causa
subyacente. Por el contrario, la recurrencia de la vasculitis después del
tratamiento puede anunciar la recaída de un tumor maligno tratado.
CARACTERÍSTICAS
CLÍNICAS
Examen físico
La vasculitis de vasos pequeños de la
piel de manera clásica se presenta con grupos de pápulas purpúricas, redondas,
de 1 a 3 mm que aparecen por más de 1 o 2 días en áreas en declive, como las
piernas. Las áreas de presión o trauma, tales como las líneas del calcetín o debajo
de los dispositivos de compresión secuencial en el paciente hospitalizado,
puede involucrarse con más fuerza. Aunque se prefieren estos sitios, las
lesiones pueden aparecer en cualquier parte, y para los pacientes confinados a
la cama, la clara preferencia por las zonas de declive pueden ser menos
aparente o alterada, y favorecer sitios como la espalda. En la vasculitis por
IgA, la púrpura palpable por encima de la cintura puede ser un marcador de
vasculitis renal, pero esto es polémico. El número de lesiones puede variar de
decenas a cientos. Nuevas lesiones pueden aparecer todos los días hasta que se
inicia el tratamiento o se retire el desencadenante;
se resuelven durante 2 a 3 semanas y poco a poco se desvanecen, y dejan marcas
de hiperpigmentación postinflamatoria.
Además de la púrpura palpable típica, también
pueden estar presentes lesiones petequiales o purpúricas más sutiles y no palpables.
Pueden desarrollarse placas más grandes o incluso úlceras como pápulas
purpúricas coalescentes (Fig. 2). Reacciones vigorosas y necrosis isquémica subsecuente
resultan en vesículas, pústulas y pequeñas bullas hemorrágicas (Fig. 3).
Pápulas tipo habón y placas pueden imitar la urticaria pero ser el signo de
presentación de la vasculitis de vasos pequeños. A diferencia de la urticaria verdadera,
estas lesiones persisten más de 24 horas, arden más que producir prurito, y
dejan marcas equimóticas en la resolución. Algunos de estos pacientes se clasifican
mejor como vasculitis urticarial, con niveles bajos o normales de complemento.
Están ausentes las manifestaciones más
típicas de vasculitis de vasos medianos, tales como nódulos subcutáneos, livedo
reticularis, púrpura retiforme, bullas hemorrágicas grandes, ulceración más
significativa y necrosis. El aspecto irregular o reticular de la piel purpúrica
alimentada por vasos de tamaño mediano de la dermis profunda o subcuticular
refleja la ramificación irregular y la distribución de los vasos pequeños más
distales. Si se ven estas lesiones, debe ser alta la sospecha de vasculitis de
vasos medianos, es decir, poliarteritis nodosa sistémica o cutánea, o para la vasculitis
crioglobulinémica o asociada a ANCA, que
puede afectar tanto a los vasos cutáneos pequeños y medianos y se presentan con
púrpura palpable.
Historia
Los pacientes con vasculitis de vasos
pequeños de la piel pueden quejarse de ardor, picazón o dolor. Pueden
experimentar edema doloroso e incómodo de las extremidades afectadas. O, pueden
estar completamente asintomáticos. Una historia cuidadosa y la revisión de los
sistemas son esenciales para la separación de los pacientes con vasculitis
limitada a la piel de aquellos con afectación sistémica significativa o con una
enfermedad subyacente. Debido a que estos pacientes pueden tener hallazgos
físicos idénticos en la presentación inicial, la historia clínica es de suma
importancia. Se debe prestar atención especial a la evidencia de vasculitis
sistémica, como fiebre, pérdida de peso y otros síntomas constitucionales;
artralgias o artritis; mialgias; dolor abdominal, melena o hematoquecia; tos,
hemoptisis, disnea o; hematuria u orina espumosa; sinusitis o rinitis; y
parestesias, debilidad o pie caído.
En la mayoría de los casos de vasculitis
de vasos pequeños de la piel, las manifestaciones sistémicas significativas son
improbables. Aunque las artralgias son bastante comunes durante las erupciones,
la sinovitis franca o artritis es rara y sugiere la presencia de enfermedad
sistémica. Si uno o más de estos síntomas están presentes, un estudio
diagnóstico objetivo debe proceder con el objetivo de identificar las
manifestaciones extracutáneas o sistémicas potencialmente graves de vasculitis
de vasos pequeños o medianos. Además de los síntomas analizados, se les debe
preguntar sobre los desencadenantes potenciales, como síntomas infecciosos
previos, ingesta de medicamentos prescritos y no prescritos, y condiciones
médicas comórbidas.
El sello distintivo de la vasculitis por
IgA, además de las manifestaciones cutáneas típicas de vasculitis de vasos
pequeños de la piel, es la presencia de síntomas gastrointestinales, tales como
dolor abdominal y sangrado gastrointestinal en 65%, artralgia o artritis con
inflamación periarticular en 63%, y la implicación renal con hematuria
microscópica o macroscópica en 40%. Debido a que los factores desencadenantes
infecciosos son comunes, la historia debe incluir información acerca de las
infecciones, en particular infecciones respiratorias superiores, en las semanas
anteriores, así como una historia médica detallada y el registro de administración
de medicamentos. Los factores que parecen predecir la afectación renal con la vasculitis
de IgA son la edad mayor de 6 años, púrpura persistente, dolor abdominal
intenso, y síntomas renales en el momento del inicio.
ESTUDIO
INICIAL
Estudios de laboratorio
Después de una historia cuidadosa, revisión
de los sistemas, y examen físico, debe proceder un estudio de laboratorio sistemático
y específico. No existe un protocolo estándar para este estudio inicial, pero
las pruebas de escrutinio deben aspirar a dilucidar la causa subyacente y la
extensión de la participación de los órganos y deben guiarse por signos y
síntomas clínicos. Cuando la presentación es sencilla, hay un medicamento
reciente o desencadenante infeccioso, y la revisión de los sistemas es negativa,
no se requiere más que un hemograma completo, pruebas metabólicas básicas y
análisis de orina (con micro). De estas pruebas, el análisis de orina es más
esencial, debido a que la presencia de glomerulonefritis es más probable que
cambie el tratamiento.
La prueba de sangre oculta en heces se
debe considerar en todos los pacientes y, ciertamente, debe realizarse si el
paciente tiene síntomas abdominales o sangrado gastrointestinal. Una
radiografía de tórax debe ordenarse si el paciente se queja de disnea, y debe
proceder otro estudio dirigido específico, si se justifica, de acuerdo a la
revisión de los sistemas y el examen físico.
Para aquellos sin una causa obvia de
vasculitis, un estudio diagnóstico inicial razonable incluye un conteo
sanguíneo completo; panel metabólico básico; análisis de orina; pruebas de
función hepática; serologías infecciosas, que incluyan hepatitis B y C, virus
de la inmunodeficiencia humana, y antiestreptolisina O; y estudio diagnóstico
reumatológico, que incluya anticuerpos antinucleares y factor reumatoide, que
puede ser como tamizaje para artritis reumatoide y un sustituto para la presencia
de crioglobulinas mixtas. Las pruebas de segundo nivel pueden incluir
electroforesis de proteínas del suero con inmunofijación para buscar evidencia
de una paraproteína; niveles séricos del complemento C3 y C4, que pueden ser
bajos en el contexto de la vasculitis urticarial o lupus sistémico y sugerir
afectación sistémica más significativa; ANCA, que son muy sugestivos de
vasculitis asociada a ANCA, si son positivos considerar crioglobulinas.
La vasculitis por hipersensibilidad es
relativamente menos común en los niños en comparación con los adultos, mientras
que la vasculitis por IgA es mucho más común. Con este fin, un estudio diagnóstico
inicial más limitado es apropiado en los niños para ir junto con la historia y
el examen, como hemograma completo, pruebas metabólicas básicas, análisis de
orina, prueba de sangre oculta en heces, y títulos de antiestreptolisina O. Por
el contrario, dada la preocupación por la afectación sistémica con vasculitis
por IgA, se debe dar especial atención a estos estudios. El valor de
laboratorio más comúnmente anormal en la vasculitis de vasos pequeños de la
piel es una velocidad de sedimentación globular elevada.
Hallazgos histológicos
Una biopsia de piel se debe realizar
siempre que sea posible para confirmar el diagnóstico y orientar aún más el
abordaje; incluso el clínico más astuto puede ser engañado por las condiciones
que imitan una vasculitis de vasos pequeños.
DIAGNÓSTICO
DIFERENCIAL
- Vasculitis crioglobulinémica
- Vasculitis asociada a ANCA
- Mordeduras de artrópodos
- Púrpura macular debida a un
traumatismo, fragilidad de la piel, o anticoagulación
- Disfunción o deficiencia plaquetaria
- Dermatosis purpúricas pigmentadas
- Émbolos de colesterol
- Émbolos sépticos
- Vasculopatía livedoide
La morfología de la lesión determina el
tipo de biopsia realizada. Para manifestaciones de vasculitis de vasos
pequeños, una biopsia en sacabocados debe ser suficiente para analizar toda la
dermis. Debido a la progresión dinámica natural del depósito de complejos
inmunes a infiltración inflamatoria, ruptura de vasos, trombosis, necrosis, y
curación; el momento y el lugar de la biopsia son críticos. Una biopsia realizada
demasiado pronto o tarde puede no ser diagnóstica. De manera ideal, una lesión representativa
debe tomarse cuando es relativamente “fresca”, es decir, aproximadamente 24 a
48 horas de aparición. Debe hacerse todo lo posible para llevar a cabo una
biopsia el mismo día en que se ve al paciente, y más si hay nuevas lesiones. Si
están presentes lesiones “más profundas”, como nódulos subcutáneos o púrpura
retiforme, debe realizarse una biopsia
por punción o una cuña adecuadamente profunda para visualizar los vasos
medianos y descartar la presencia de una vasculitis de vasos medianos o pequeños
a medianos.
Los hallazgos prototípicos de la vasculitis
leucocitoclástica incluyen una infiltración neutrofílica de los vasos
sanguíneos pequeños en la dermis superficial y media, los desechos granulocíticos
y el polvo nuclear (leucocitoclasia), necrosis fibrinoide e interrupción de las
paredes del vaso, y extravasación de las células rojas de la sangre en el tejido
circundante. Un infiltrado inflamatorio mixto también puede estar presente,
sobre todo en lesiones mayores. La presencia de eosinofilia tisular sugiere que
la vasculitis puede ser inducida por medicamentos. Algunos sugieren que la
gravedad de los cambios histológicos y la profundidad del infiltrado
inflamatorio pueden predecir la gravedad de la enfermedad y la afectación
sistémica o incluso la presencia de malignidad subyacente. La presencia de
vasculitis granulomatosa es sugestiva de la granulomatosis con poliangeítis o
la granulomatosis eosinofílica con poliangeítis.
Es importante tener en cuenta que estos
patrones histológicos pueden verse en cualquier síndrome vasculítico y no son
específicos para cualquier entidad particular. Las infecciones, reacciones a
mordeduras de insectos, dermatosis neutrofílica (síndrome de Sweet, pioderma
gangrenoso) y úlceras debidas a otras causas pueden exhibir una vasculitis
leucocitoclástica secundaria al proceso primario. La correlación
clínico-patológica, como siempre, es necesaria antes de decidirse por un
diagnóstico final.
Siempre que sea posible, debe realizarse una
segunda biopsia para estudios de inmunofluorescencia directa. La detección de
depósitos de complejos inmunes puede tener significancia pronóstica. La
presencia de depósitos de IgA en las paredes del vaso es la característica
patológica definitoria de la vasculitis por IgA y aumenta la probabilidad de los
síntomas renales y gastrointestinales que se ven en ese síndrome. Mientras
tanto, la presencia de IgM en la piel lesionada puede correlacionar con
involucro renal o crioglobulinemia. Una banda continua de C3 o IgG en la unión
dermoepidérmica (prueba de banda positiva para lupus) puede sugerir vasculitis
urticarial hipocomplementémica y lupus eritematoso sistémico subyacente.
La selección adecuada del sitio de la
biopsia es de nuevo crítica, debido a que los complejos inmunes son más
propensos a verse en las lesiones tempranas entre 8 y 24 horas de edad. Debido
a que la cascada inflamatoria posterior destruye los complejos inmunes, las
lesiones mayores pueden ser falsamente negativas. Las biopsias para inmunofluorescencia
directa se deben tomar de la piel lesionada, y la muestra se debe colocar en el
medio de Michel o solución salina normal para su procesamiento.
Por último, el término “vasculitis
leucocitoclástica” a veces se utiliza de manera incorrecta en los informes
histológicos para describir infiltrados perivasculares aislados que carecen de
los infiltrados de la pared de los vasos o necrosis fibrinoide. Aunque esos
especímenes pueden, en verdad, representar vasculitis temprana de vasos pequeños,
no son diagnósticos de la enfermedad y pueden dar lugar a confusión. Por tanto,
es importante leer el informe de patología en detalle, como el estudio de
inmunofluorescencia directa, si se hace, y no confiar sólo en el diagnóstico impreso.
Tratamiento inicial
Una vez que se realizó un diagnóstico de
vasculitis de vasos pequeños en la piel, el paso inicial más importante en el
abordaje es, como se indica, la evaluación para su posible afectación
sistémica, seguida del tratamiento para condiciones subyacentes, según lo
dictado por el examen, la historia, y la revisión de los sistemas. Los
resultados de estas investigaciones dictan el tratamiento inicial, debido a que
es necesaria terapia sistémica más agresiva en el caso de involucro renal o de
otros órganos, y el cese de un agente causal o el tratamiento de una condición
subyacente se indican si se identifica uno.
Una vez que se excluyó afectación
sistémica, el tratamiento de la vasculitis de vasos pequeños de la piel debe
centrarse en el tratamiento de los síntomas. Debido a que la mayoría de los
casos afectan sólo la piel, son agudos y autolimitados, en general no es
aconsejable terapia inmunosupresora agresiva. Los desencadenantes
identificables deben eliminarse o tratarse. Se debe evitar permanecer mucho
tiempo de pie. El descanso y la elevación y el uso de medias de compresión
pueden ser útiles para disminuir el depósito de complejos inmunes relacionado
con la estasis y acelerar la curación de las úlceras en las piernas. Los
esteroides tópicos pueden aliviar el prurito o el ardor, pero no prevenir
lesiones nuevas. Más de la mitad de los pacientes no requieren tratamiento
sistémico.
Tratamiento subsecuente
Las terapias sistémicas se indican para
una enfermedad grave, intratable, o recurrente. El malestar asociado, la ulceración
y el impacto psicosocial justifican una intervención. Cualquier episodio
inicial que no se autolimita y que dura más que unas pocas semanas
probablemente requiera tratamiento sistémico, incluso si es relativamente
asintomático. Por desgracia, hay una escasez de datos de alta calidad para el
tratamiento directo, y más de 1 agente es a menudo probado antes de alcanzar el
éxito. Para los pacientes con vasculitis crónica de vasos pequeños de la piel,
la resolución completa o la curación pueden permanecer difíciles de alcanzar,
pero a menudo los medicamentos antiinflamatorios trabajan sin la necesidad de
usar glucocorticoides sistémicos.
De manera inicial, las opciones de
tratamiento a largo plazo incluyen la colchicina (0.6 mg 2-3 veces al día, si
es tolerada). Este medicamento se reporta útil para la piel y los síntomas
articulares en estudios abiertos, e induce pronta resolución de la vasculitis
cutánea. A pesar de que no mostró beneficios significativos en un ensayo
aleatorizado y controlado de 20 pacientes (el único estudio de este tipo para vasculitis
cutáneas de vasos pequeños), los pacientes incluidos fueron refractarios a
otras terapias y un subconjunto de ellos respondió y se exacerbó cuando tomaron
la medicación. La dosificación puede estar limitada por los efectos secundarios
gastrointestinales.
El uso de dapsona (50 a 200 mg/d), que
tiene efectos antineutrofílicos, se apoyó en la experiencia anecdótica y series
pequeñas de casos. Se contraindica en pacientes con deficiencia de
glucosa-6-fosfato deshidrogenasa, y puede causar metahemoglobinemia y anemia
por hemólisis, lo que exige un monitoreo regular de laboratorio. Se puede
combinar con colchicina cuando es incompleta la respuesta a la monoterapia con
cualquiera de estos medicamentos. Otras opciones incluyen hidroxicloroquina
(200-400 mg/d), que es útil principalmente en la vasculitis urticarial; pentoxifilina
(400-1200 mg/d), que también se puede combinar con dapsona; y agentes antiinflamatorios
no esteroideos, como la indometacina, que pueden ayudar a aliviar los síntomas.
El uso de estos agentes sólo es compatible con series de casos y datos
anecdóticos.
Los corticoesteroides sistémicos, con
dosis iniciales de 0.5 a 1 mg/kg por día de prednisona, se recomiendan para las
personas con lesiones necróticas graves o graves manifestaciones sistémicas,
como afectación renal o gastrointestinal. La respuesta a este tipo de
tratamiento suele ser rápida, y la dosis debe reducirse de forma lenta para
evitar el rebote. El tratamiento a largo plazo puede no ser necesario si el
proceso se autolimita. Por desgracia, en los casos de vasculitis cutánea
crónica, los esteroides sistémicos, con sus efectos secundarios asociados, no
son una buena opción a largo plazo. En una revisión de Cochrane de ensayos
controlados aleatorizados, el tratamiento temprano con corticoesteroides en el
momento del diagnóstico no mostró ningún beneficio para la prevención de
complicaciones renales de la vasculitis por IgA. Debido a que los corticoesteroides
no parecen prevenir las complicaciones renales de la vasculitis por IgA, no se
recomienda su uso profiláctico. Sin embargo, lo que hacen es aliviar el dolor
articular y abdominal y parecen acelerar la resolución de los síntomas renales.
En los pacientes con síntomas renales graves y en aquellos que desarrollan
enfermedad renal, debe considerarse su uso.
Cuando ninguno de estos agentes es eficaz
o tolerado, o si la vasculitis es crónica o refractaria, pueden considerarse medicamentos
inmunosupresores como azatioprina (de 50-200 mg/d, si los niveles de tiopurina
metiltransferasa son normales), metotrexato (15-25 mg/semana), con suplementos
de ácido fólico, y micofenolato mofetil (2.3 g/d), y equilibrar los riesgos de
estos agentes en contra de la gravedad de la condición de la piel y si las manifestaciones
sistémicas están presentes. Los agentes más fuertes y más tóxicos como
ciclofosfamida pueden ser eficaces. Los inhibidores del factor de necrosis
tumoral, en especial el infliximab, pueden ser eficaces para la vasculitis de
vasos pequeños de la piel, pero también pueden causar vasculitis. El rituximab
se utilizó en algunos casos refractarios.
Pronóstico
La mayoría de los episodios de vasculitis
de vasos pequeños de la piel se autolimitan, pueden resolver de 3 a 4 semanas
con hiperpigmentación residual y no se repiten. En general, la afectación
sistémica (si se encuentra) es mínima. Sin embargo, la disfunción grave de
órganos internos ocurre rara vez. El pronóstico depende de la gravedad de la
afectación de órganos, así como cualquier trastorno médico subyacente asociado.
Un cierto número de pacientes, tal vez 8% a 10%, puede desarrollar vasculitis
crónica o recurrente. A pesar de esto, la mayoría de los casos tienen un buen
pronóstico global.
La duración habitual de la vasculitis por
IgA es entre varias semanas a algunos meses (promedio de 4 semanas). Ninguna
terapia demostró acortar la duración de la enfermedad. Hasta un tercio de los
pacientes tienen enfermedad persistente o recurrente de hasta 6 meses. El seguimiento
cercano, con la repetición frecuente de los análisis de orina y el control de
la presión arterial, se debe continuar por lo menos ese tiempo. El pronóstico
de la vasculitis por IgA es en general favorable, pero depende principalmente
de la gravedad de la enfermedad renal. Incluso los niños con hematuria
macroscópica por lo general se recuperan. Sin embargo, la enfermedad glomerular
progresiva y la insuficiencia renal pueden ocurrir en 1% a 3%, por lo que los
pacientes con hematuria o proteinuria deben seguirse de manera cuidadosa para
prevenir el deterioro de la función renal. La insuficiencia renal y las secuelas
a largo plazo son más comunes en los adultos. Aún así, la nefropatía
persistente, generalmente leve ocurre en sólo 8% de los pacientes adultos.
RESUMEN
A pesar del buen pronóstico en general de
la vasculitis de vasos pequeños de la piel, este grupo de condiciones puede ser
una fuente importante de morbilidad, en especial para aquellas personas que
padecen una enfermedad sistémica o crónica/recurrente. La falta de datos de
alta calidad para guiar el diagnóstico, el tratamiento y el seguimiento de la
vasculitis cutánea es un área continua de necesidad. Son necesarias series más
grandes con seguimientos sustanciales y bien diseñados y ensayos terapéuticos
controlados para ayudar a desarrollar guías basadas en la evidencia para la
evaluación y el tratamiento de estas condiciones.



Centro Regional de Alergia e Inmunología
Clínica CRAIC, Hospital Universitario “Dr. José
Eleuterio González” UANL, Monterrey, México
Dra. med. Sandra Nora González Díaz Jefe y Profesor
Dr. José Antonio Buenfil López Profesor
Dr. Carlos Macouzet Sánchez Residente 1er Año
Dra. Alejandra Macías Weinmann Profesor



No hay comentarios:
Publicar un comentario
Nota: solo los miembros de este blog pueden publicar comentarios.