La vasculitis sistémica denota la inflamación de forma patológica de un vaso sanguíneo, que se caracteriza por la presencia de un infiltrado inflamatorio y destrucción de la pared del vaso, lo que provoca estenosis y trombosis. La vasculitis es un grupo de diversos trastornos que demuestran la afectación de varios órganos y la gravedad clínica. La vasculitis puede afectar cualquier vaso en el sistema de órganos y, según el vaso al que invada, las manifestaciones pueden ser muy diversas. Es muy común que afecciones muy variables provoquen retrasos en el diagnóstico. Por lo tanto, la identificación temprana de la vasculitis, la evaluación de la respuesta al tratamiento y la detección de la recaída de la enfermedad son desafíos clínicos importantes.
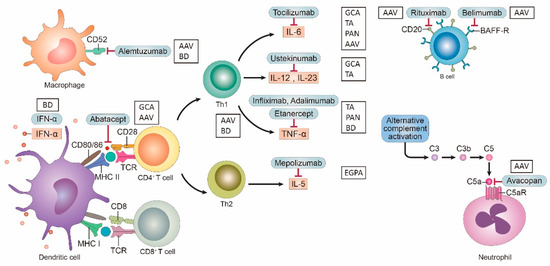
La patogenia de la vasculitis sigue sin estar clara. Una explicación es que la exposición a un antígeno sin identificar como un virus, una toxina o un epítopo críptico, conduce a la activación de la respuesta inmune. En algunas personas, esta respuesta inmunitaria no disminuye su expresión, lo que lleva a la producción de complejos inmunes que se depositan en las paredes de los vasos sanguíneos y provocan vasculitis. Las vasculitis pauciinmunes no están mediadas por inmunocomplejos y de forma típica se asocian con autoanticuerpos anticitoplasma de neutrófilos (ANCA), que se supone que causan daño vascular de modo indirecto al inducir a los neutrófilos a desgranularse y producir radicales libres de oxígeno.
La última década fue testigo de avances importantes en la comprensión de la patogenia de las vasculitis. Estos descubrimientos llevaron al desarrollo de nuevos tratamientos, que buscan proporcionar una mayor eficacia y un perfil más aceptable de efectos secundarios. En esta revisión, se discuten los avances recientes en la comprensión de la patogenia de las vasculitis sistémicas primarias y el desarrollo de nuevos tratamientos.
2. Clasificación de vasculitis sistémica
Los criterios de clasificación se destinan a crear grupos de pacientes homogéneos para la investigación. Los sistemas de clasificación de las vasculitis se limitan por las características superpuestas de los subgrupos y los mecanismos patogénicos no reconocidos. Los criterios de clasificación más utilizados se definen por el tamaño del vaso al que afectan de manera predominante, es decir, pequeño, mediano o grande, o tamaño variable del vaso (Tabla1). La Conferencia de Consenso Internacional de Chapel Hill (CHCC) de 2012 definió y estandarizó la nomenclatura de las vasculitis sistémicas.
Las VVG involucran la aorta y sus ramas principales, e incluyen la arteritis de células gigantes (ACG) y la arteritis de Takayasu (AT). Las VVM involucran las principales arterias y venas viscerales y sus ramas iniciales e incluyen la poliarteritis nodosa (PAN) en adultos. La enfermedad de Kawasaki, la otra forma importante de VVM y una arteritis aguda de la infancia, no se trata en esta revisión. Las VVP involucran arteriolas, capilares, arterias intraparenquimatosas, vénulas y algunas venas e incluyen la vasculitis asociada a ANCA (VAA), la VVP más común en adultos. Sin embargo, existe cierta superposición, y las arterias de cualquier tamaño pueden involucrarse de forma potencial en cualquier caso de las tres categorías principales de afectación del patrón de vasos dominante. Además de las vasculitis sistémicas multiorgánicas, también se definen otras formas de vasculitis, tales como vasculitis de un solo órgano, que incluyen la arteritis cutánea, la vasculitis primaria del sistema nervioso central y la aortitis aislada; la vasculitis asociada con enfermedad sistémica, que incluye la vasculitis reumatoide, la vasculitis lúpica y la vasculitis sarcoide; y la vasculitis con asociación a una causa subyacente: relacionada con enfermedad (hepatitis B, crioglobulinemia asociada a hepatitis C y cáncer), o la vasculitis con asociación a fármacos.
La característica central de la VVG es la arteritis granulomatosa. La ACG afecta de manera exclusiva a personas > 50 años con un predominio de mujeres a hombres de 3: 1. Además, la ACG es más común en pacientes de ascendencia del norte de Europa que en grupos étnicos asiáticos. La ACG afecta de forma típica a las ramas de las arterias carótida, vertebral y temporal, lo que da como resultado los síntomas clásicos de dolor de cabeza, claudicación mandibular y pérdida de visión. Por el contrario, la AT suele afectar a las mujeres durante la segunda y tercera décadas de la vida. Es raro en el norte de Europa, pero es más común en el sudeste asiático. La AT afecta de forma típica a la aorta y sus ramas primarias, lo que lleva a la oclusión vascular con claudicación, formación de aneurismas, insuficiencia aórtica e insuficiencia cardíaca. El tratamiento actual de la VVG son glucocorticoides. Aunque el metotrexato y la azatioprina se utilizaron como agentes ahorradores de esteroides, su eficacia no se demostró en ensayos controlados aleatorios (ECA).
La PAN es poco común, con una incidencia que se estima de 1 a 10 por millón. Ambos sexos se afectan por igual y la edad máxima de aparición es entre los 40 y los 60 años. La etiopatogenia de la PAN se relaciona de forma estrecha con la infección por hepatitis viral, en particular el virus de la hepatitis B, que comprendió más de un tercio de los 348 casos de PAN en la serie de casos más grande hasta la fecha. Cerca de 35% de los casos de poliarteritis nodosa (PAN) se asocian con la hepatitis B. La incidencia de PAN que se relaciona con el virus de la hepatitis B disminuyó de manera sustancial durante las últimas cuatro décadas después de las mejoras en la inmunización, la práctica de transfusiones y la terapia para el virus de la hepatitis B. La PAN se caracteriza por una arteritis necrotizante transmural de las arterias musculares. Los sitios más afectados de forma común son la piel (que causa livedo reticularis y ulceración) y los nervios periféricos (que provocan una mononeuritis múltiple). La afectación de los vasos viscerales también es común con múltiples estenosis arteriales irregulares y microaneurismas demostrables en la angiografía con contraste en hasta 90% de los pacientes con inmunosupresión a largo plazo con glucocorticoides junto con otros agentes como ciclofosfamida, metotrexato o azatioprina, mejora los resultados del paciente y apoya un componente autoinmune de la patogénesis.
Las vasculitis asociadas a anticuerpos (VAA) anticitoplasma de neutrófilos (ANCA) son un grupo de trastornos autoinmunitarios sistémicos que afectan de forma predominante a los vasos pequeños. Las VAA son vasculitis necrotizantes que se diferencian de otras vasculitis de vasos pequeños por la falta de un depósito inmunitario significativo en las paredes de los vasos. La VAA incluye la poliangeítis microscópica (MPA), la granulomatosis con poliangeítis (GPA) y la granulomatosis eosinofílica con poliangeítis (GEPA). Los autoanticuerpos que definen la VAA son mieloperoxidasa (MPO)-ANCA y proteinasa 3 (PR3)-ANCA. Las VAA son enfermedades autoinmunes poco frecuentes con una prevalencia combinada estimada de 46-184 por millón. Sin embargo, se asocian con una mortalidad significativa. Se reporta que la mortalidad por GPA es superior a 90% a los dos años si no se trata. De manera afortunada, la introducción de terapias efectivas redujo de manera drástica la tasa de mortalidad a dos años a 15%. El tratamiento de la VAA consiste en la inducción a la remisión seguida de una fase de mantenimiento. Las recomendaciones de tratamiento para la inducción o el mantenimiento de la VAA varían según la gravedad de la enfermedad. Existen diferentes definiciones para determinar qué constituye una enfermedad grave, pero, en general, se considera grave cualquier VAA que ponga en peligro la vida o los órganos. Las opciones de tratamiento actuales son efectivas; sin embargo, se asocian con una morbilidad significativa del paciente debido a los efectos adversos relacionados con el tratamiento.
La enfermedad de Behçet (EB) es una vasculitis multisistémica recidivante rara, caracterizada por ataques recurrentes de úlceras genitales, orales y afectación ocular, musculoesquelética, vascular, del sistema nervioso central (SNC) y gastrointestinal (GI). La prevalencia de EB varía de forma amplia según el área geográfica, pero, según un metaanálisis reciente, es cercana a 10.3 por 100.000 habitantes. La afectación vascular es la causa más frecuente de mortalidad y la afectación ocular es el factor de morbilidad más importante en la EB ya que puede provocar ceguera. El tratamiento de EB se basa en manifestaciones clínicas. Si bien la colchicina, los agentes antiinflamatorios no esteroideos y los tratamientos tópicos a menudo son suficientes para la afectación mucocutánea y articular, se requieren agentes inmunosupresores para la afectación de órganos importantes.
A medida que aumenta la comprensión de la fisiopatología de la vasculitis sistémica, se proponen nuevas terapias con menos efectos tóxicos. Este artículo proporcionará una revisión de las opciones de tratamiento actuales y una opinión experta sobre el futuro del tratamiento de la VAA.
3. Descubrimiento de fármacos y dianas potenciales en la vasculitis
El tratamiento de los diversos tipos de vasculitis se basa de forma principal en corticoesteroides y fármacos inmunosupresores convencionales, como metotrexato o azatioprinas. Dado que la vasculitis es una enfermedad inflamatoria crónica compleja, es posible que se necesite tratamiento para muchas moléculas y dianas inflamatorias diferentes. En la actualidad, la investigación de estas moléculas y sus dianas se basa de manera principal en algunos anticuerpos o inhibidores. Los avances recientes en la era de los agentes biológicos mejoran de manera drástica el manejo de los casos difíciles de tratar. (Tabla 2, Figura 1).
3.1. Citocinas Th1 y el descubrimiento de los medicamentos correspondientes
3.1.1 IL-6
La IL-6 tiene un efecto patológico sobre la respuesta inflamatoria tanto en la pared del vaso como en la circulación sistémica. El tocilizumab es un anticuerpo monoclonal humanizado que inhibe de forma competitiva la IL-6 al unirse a los receptores de la IL-6 circulantes y unidos a la membrana. El primer ensayo controlado aleatorizado que reportó sobre la eficacia de tocilizumab en ACG asignó al azar a 20 pacientes a 8 mg/kg de tocilizumab administrado por vía intravenosa cada mes o infusiones de placebo además de glucocorticoides y encontró una mayor supervivencia libre de recaídas en el grupo de tocilizumab (85 versus 20 %, p = 0.001) en la semana 52. Se observaron efectos de tocilizumab sobre el ahorro de glucocorticoides tanto en la ACG recidivante como en la recién diagnosticada. El ensayo fase 3 Arteritis de Células Gigantes Actemra (GiACTA) reclutó a 251 pacientes con ACG de nueva aparición, asignados al azar a uno de cuatro brazos: tocilizumab 162 mg por semana o en semanas alternas (combinado con una disminución gradual de prednisona de 26 semanas) o una disminución gradual de prednisona sola (26 o 52 semanas). Este estudio reportó que el tocilizumab es una terapia eficaz que ahorra glucocorticoides, que demuestra una remisión sostenida sin glucocorticoides en 56% de los pacientes que reciben tocilizumab cada semana en comparación con 18% de los pacientes que reciben una reducción gradual de prednisona durante 52 semanas. El tocilizumab se aprobó por la Administración de Alimentos y Medicamentos (FDA) para el tratamiento de la ACG.
En la AT, en 2017 se reportó un ensayo fase 3 sobre el efecto del tocilizumab en la arteritis de Takayasu (TAKT). Aquí, 36 pacientes con AT recidivante se asignaron al azar a tocilizumab, 162 mg semanales o placebo administrados de forma semanal junto con una dosis decreciente de glucocorticoides. Al analizarse mediante un método de intención de tratar, el tocilizumab no mostró una diferencia en el tiempo hasta la recaída en comparación con el placebo (índice de riesgo [HR] 0.41; intervalo de confianza [IC] de 95%: 0.15-1.10, p = 0.0596). Sin embargo, el análisis por protocolo mostró una diferencia significativa para el tocilizumab (n = 16) versus placebo (n = 17) (HR 0.34; IC de 95%: 0.11-1.00, p = 0.03). En 2020, se reportó la eficacia y seguridad a largo plazo del tocilizumab en la AT. En ese estudio, 28 pacientes recibieron tocilizumab durante 96 semanas. Cuarenta y seis punto cuatro por ciento de estos 28 pacientes tratados con tocilizumab redujeron su dosis a <0.1 mg/kg/día, lo que muestra así evidencia de un efecto ahorrador de esteroides del tocilizumab en la AT en el tratamiento a largo plazo.
Todavía no hay ningún ECA sobre el efecto del tocilizumab en la PAN. En un reporte reciente de caso, el tocilizumab fue eficaz para la PAN relacionada con el virus de la hepatitis B sin reactivación del virus de la hepatitis B. En una revisión de la literatura basada en 11 reportes de casos, el tocilizumab fue eficaz en casos de poliarteritis nodosa refractaria o recidivante y mostró su efecto de ahorro de glucocorticoides.
Hay varios reportes de casos que describen pacientes con VAA tratados con tocilizumab que muestran que se logró una remisión completa y sostenida en muchos de los pacientes con enfermedad refractaria. Los ECA pueden justificarse en el futuro.
3.1.2. IL-12 e IL-23
La IL-23 es una citocina proinflamatoria compuesta por dos subunidades, IL-23A (p19) e IL-23B (p40), esta última compartida con IL-12. El eje IL-23/IL-17 ejerce de forma principal un papel protector contra las infecciones bacterianas; su desregulación desempeña un papel en los trastornos inflamatorios inmunomediados. Como se reportó que la vía IL-12/células Th1/IFN-γ se involucra en la inflamación granulomatosa en la patogénesis de la ACG, se intentaron tratamientos dirigidos a IL12 y se reportó el uso de ustekinumab para tratar la VVG.
El ustekinumab es un anticuerpo monoclonal que se dirige a la subunidad p40 de IL-12/23. Un estudio abierto de 25 pacientes con ACG refractaria tratados con ustekinumab además de glucocorticoides demostró que ningún paciente recayó durante 52 semanas. La dosis media de prednisolona disminuyó de 20 a 5 mg y cerca de 25% de los pacientes pudieron suspender los glucocorticoides. Además, la angiografía por TC mostró una mejora en el grosor de la pared con resolución completa en ocho pacientes que se sometieron a una angiografía por TC antes y después del tratamiento. Sin embargo, en un estudio prospectivo reportado de forma reciente, 10 de 13 (77%) pacientes no lograron el criterio principal de valoración con ustekinumab en la reducción gradual de prednisona, y siete experimentaron brotes de enfermedad después de un período promedio de 23 semanas. Parece justificado realizar más investigaciones sobre el efecto de ustekinumab en la ACG.
El tratamiento con ustekinumab en la AT se reportó de forma esporádica y sólo en unas pocas series de casos. Una serie de tres pacientes con AT refractaria tratados con ustekinumab reportó estabilización de la actividad clínica de la enfermedad y normalización de marcadores inflamatorios. De manera reciente, los resultados de un seguimiento a largo plazo de estos tres pacientes reportaron que el ustekinumab mostró efectos marginales en la reducción de la dosis de prednisolona, y 2 de 3 pacientes interrumpieron el tratamiento con ustekinumab debido a una recaída y un fracaso secundario.
3.1.3. Inhibidor del factor de necrosis tumoral (TNF) α
Los inhibidores del TNF α fueron los primeros agentes biológicos probados en diversas vasculitis. El TNF α es una citocina importante para la formación de granulomas, y también para la activación de células endoteliales. Después de que se reportaron algunos casos que mostraban un tratamiento exitoso con anti-TNF-α en pacientes con ACG, se intentó un estudio comparativo doble ciego que utilizó infliximab, pero de forma posterior se detuvo debido a la falta de eficacia en la prevención de recaídas. Con el uso del etanercept, un estudio controlado aleatorizado mostró un efecto significativo de ahorro de esteroides después de un año en 17 pacientes, sin embargo, no durante un período más largo. La adición de adalimumab a un régimen estándar de prednisona no mostró ningún efecto ahorrador de esteroides en 70 pacientes en un estudio controlado aleatorizado prospectivo de 10 semanas. Como resultado de estos estudios, no se recomienda la terapia anti-TNF-α en la ACG. Varios estudios retrospectivos y series de casos reportaron que los inhibidores del TNF α fueron efectivos en la mayoría de los pacientes con arteritis de Takayasu refractaria. Un estudio de cohorte de seguimiento de dos años de Noruega reportó tasas más altas de remisión sostenida, así como menor progresión de las lesiones angiográficas en pacientes que reciben agentes anti-TNF-α, en comparación con los tratamientos convencionales en la arteritis de Takayasu.
En la PAN, el infliximab se utilizó en formas refractarias de la enfermedad o por intolerancia a los fármacos convencionales y parece ser eficaz. En una serie pequeña de casos, nueve pacientes refractarios con PAN se trataron con infliximab y 8 de 9 pacientes (89%) lograron una mejoría significativa y una reducción de la dosis de prednisona de 50%.
Varios estudios abiertos y series de casos reportaron de la utilidad de las terapias anti-TNF-α en la VAA, aunque estos resultados no se confirmaron en ECA. En el Ensayo Etanercept para la Granulomatosis de Wegener (WGET), que reclutó a 174 pacientes con GPA, no hubo beneficio del etanercept en la tasa de remisión sostenida. Con poca evidencia de su efectividad, el uso del tratamiento anti-TNF-α en la VAA puede limitarse de manera significativa en el futuro. De manera similar, para GEPA, la única información disponible se deriva de cinco reportes de casos con hallazgos contradictorios que no apoyan el uso de anti-TNF-α para tratar la GEPA. En un pequeño ECA de 40 pacientes con EB, el etanercept fue más eficaz de forma significativa en la supresión de la mayoría de las manifestaciones mucocutáneas, como úlceras orales y eritema nudoso, que el placebo. Varios estudios observacionales y series de casos también confirmaron los efectos beneficiosos del infliximab y el adalimumab sobre las lesiones mucocutáneas de la EB. La mayoría de los estudios sobre los efectos de los inhibidores de TNF-α en la EB se encuentran en manifestaciones oculares y se reportan de forma principal en series de casos. El infliximab disminuye de forma significativa la tasa de recaída y la dosis de glucocorticoides en pacientes con EB con afectación ocular. En el primer estudio prospectivo en 63 pacientes con uveítis por EB, la uveorretinitis mejoró con el tratamiento con infliximab en 92% y se mantuvo hasta 12 meses. Un estudio multicéntrico observacional de 1 año reportó los resultados del uso de infliximab y adalimumab en 124 pacientes con uveítis por EB refractaria, y se logró la remisión completa en 84/124 (68%). Un estudio observacional retrospectivo reciente también reportó que el adalimumab fue muy efectivo y seguro para el tratamiento de la uveítis relacionada con la EB. Un estudio abierto de 177 pacientes con uveítis relacionada con EB comparó la eficacia de infliximab (103 pacientes) versus adalimumab (74 pacientes) como agente biológico de primera línea. En este estudio, se observó una mejora en todos los parámetros oculares en ambos grupos después de 1 año de tratamiento; sin embargo, el adalimumab tuvo mejores resultados oculares de forma significativa en algunos parámetros. En las manifestaciones vasculares relacionadas con la EB, como trombosis venosa profunda, tromboflebitis superficial, un estudio retrospectivo reportó que el adalimumab logró una respuesta vascular significativa y mayor (34/35, 97%) en comparación con los inmunosupresores convencionales (23/35, 66%) durante un seguimiento de 25.7± 23.2 meses. También se observó una recaída vascular menor de forma significativa en el grupo de adalimumab. En dos estudios observacionales multicéntricos recientes, se logró la remisión clínica en 89% y 80% de los pacientes con EB, de manera respectiva, con afectación vascular refractaria al tratamiento convencional con IS.
3.2. Citocinas Th2 y descubrimiento de fármacos relativos
3.3. IL-5
La IL-5 es la citocina principal responsable de la activación, la quimioatracción y la supervivencia de los eosinófilos. Varios estudios reportaron niveles séricos elevados de IL-5 en la GEPA. De manera reciente, se estudió a los antagonistas de IL-5 como un tratamiento específico de la GEPA. El mepolizumab, un anticuerpo monoclonal anti-IL-5 humanizado, inhibe de manera selectiva la inflamación eosinofílica y se aprobó para tratar el asma eosinofílica grave. Se observó un beneficio del tratamiento con mepolizumab en pacientes con GEPA en estudios piloto abiertos pequeños previos. En el ensayo controlado aleatorizado a gran escala, 136 pacientes con GEPA con una enfermedad no controlada se inyectaron 300 mg de mepolizumab una vez al mes por vía subcutánea. La tasa de remisión en los pacientes tratados con mepolizumab fue significativa y mayor que la de los pacientes tratados con placebo (28% contra 3%, de los participantes tuvieron ≥24 semanas de remisión acumulada; razón de momios, 5.91; intervalo de confianza [IC] de 95%, 2.68 a 13.03; p < 0.001). Este estudio condujo a la autorización de la FDA para el mepolizumab como el primer fármaco aprobado de forma específica para la GEPA.
3.3. Dianas y descubrimiento de fármacos de células B
3.3.1. CD20
Las células B son fundamentales de forma clara para la patogenia de la VAA, ya que producen ANCA. El rituximab es un anticuerpo monoclonal quimérico que induce la disminución de las células B al unirse a las células B que expresan CD20. Su desarrollo dio lugar a avances en la terapia para la VAA. Los ensayos del rituximab en la vasculitis asociada a ANCA (RAVE) y el rituximab versus la ciclofosfamida en la vasculitis asociada a ANCA (RITUXVAS) establecieron la no inferioridad del rituximab a la ciclofosfamida para la inducción de la remisión de la VAA. Se desarrollaron varios fármacos anti-CD20 de segunda generación, uno de los cuales, el ofatumumab, se probó en una serie pequeña de casos de pacientes con VAA, y los resultados muestran su beneficio terapéutico. Sin embargo, todavía no hay ningún ECA.
3.3.2. BAFF
El factor de activación de células B (BAFF), también conocido como estimulador de linfocitos B (BlyS), desempeña un papel importante en la maduración de las células B y se reconoce cada vez más como importante en la patogenia de la VAA recidivante. El aumento de la expresión de BAFF es evidente en pacientes con vasculitis activa, y los datos preclínicos sugieren que concentraciones altas de BAFF pueden promover la supervivencia de las células B autorreactivas que, en condiciones normales, se degradarían. El belimumab es un anticuerpo monoclonal humanizado de forma completa que se une a los receptores BAFF en las células B. Se autorizó para el tratamiento del lupus eritematoso sistémico.
En la VAA, el ensayo de Belimumab en la Remisión de la Vasculitis (BREVAS) examinó la adición de belimumab a azatioprina y glucocorticoides para el mantenimiento de la remisión en pacientes con GPA y MPA. Sin embargo, el ensayo se detuvo antes de tiempo debido a un reclutamiento subóptimo y no se observó ninguna mejora en la tasa de recaída. La combinación de rituximab y belimumab se investiga más a fondo en un ensayo aleatorio doble ciego controlado con placebo en curso, el ensayo combinado de Rituximab y Belimumab Terapia en PR3-VAA (COMBIVAS) (identificador ClinicalTrials.gov: NCT03967925).
3.4. Coestimulación y disminución de células B y células T
3.4.1. CD28–CD80/CD86
Las moléculas coestimuladoras modulan de manera predominante las respuestas inmunitarias que activan las funciones de las células T y B, pero también afectan las funciones de las células dendríticas y los macrófagos y desempeñan un papel crucial en la inflamación. El abatacept, una proteína de fusión del dominio extracelular de CTLA-4 y el fragmento Fc de la IgG1 humana (CTLA-4-Ig), es un inhibidor de la activación de los linfocitos T mediante bloqueo coestimulador al unirse a los receptores CD80 y CD86 en las CPA que se necesitan para la activación de la presentación de antígenos de las células T.
En la ACG, se aleatorizaron 41 pacientes y la supervivencia sin recaídas fue 48% con abatacept como terapia de mantenimiento en comparación con 31% con placebo (p = 0.049). La duración de la remisión fue mayor de forma significativa en el grupo de abatacept que en el grupo de placebo. En un estudio multicéntrico controlado, aleatorizado, doble ciego, 34 pacientes con AT se trataron con abatacept a una dosis de 10 mg/kg los días 1, 15, 29 y a las 8 semanas. Los pacientes que alcanzaron la remisión a las 12 semanas se asignaron al azar para recibir placebo (n = 15) o abatacept mensual (n = 11) y seguimiento hasta los 12 meses. Sin embargo, no hubo diferencia en la duración de la remisión y la supervivencia sin recaídas a los 12 meses entre los dos grupos. En la VAA, se observaron infiltraciones de células T granulomatosas en pulmones y riñones, lo que sugiere un papel patógeno de las células T. En un ensayo abierto de 20 pacientes con GPA recidivante no grave que se trataron con abatacept, la tasa de remisión fue cerca de 80% y la tasa de interrupción de esteroides de 75%. El ensayo fase III aleatorizado controlado con placebo en curso Abatacept para el Tratamiento de Granulomatosis no Grave con Poliangeítis Recurrente (ABROGATE) (ClinicalTrials.gov identifier: NCT02108860) recluta pacientes en la actualidad.
3.4.2. CD52
El CD52 se expresa en monocitos, macrófagos y eosinófilos. El alemtuzumab puede lograr la disminución de las células B y las células T. Este anticuerpo monoclonal humanizado anti-CD52 disminuye de forma selectiva los linfocitos y se demostró que es eficaz en otras vasculitis sistémicas como la enfermedad de Behçet. El alemtuzumab como terapia de inducción de la remisión fue eficaz en 84% de 32 pacientes con EB, y se logró una remisión sostenida en 69% a los 12 meses. Walsh y colaboradores reportaron los resultados de un estudio retrospectivo a largo plazo de 71 pacientes con VAA refractaria o recidivante tratados con alemtuzumab y lo encontraron útil para lograr la remisión con una tasa de recaída más baja. Un estudio aleatorizado, prospectivo y abierto de alemtuzumab para la inducción de la remisión en VAA refractaria (ensayo ALEVIATE) se presentó en forma de resumen en 2019. En este estudio, se logró la remisión en 65% de los pacientes con VAA a los 6 meses y 35 % de remisión sostenida al año. Sin embargo, los eventos adversos, como infecciones, son altos en comparación con el tratamiento estándar.
3.6. Dianas del complemento
Receptores C5a
El sistema del complemento es un mediador central de las respuestas inmunitarias mediadas por anticuerpos. C5 es una proteína efectora potente en esta vía, que ejerce sus efectos por medio de sus productos de escisión: C5a, una potente anafilatoxina y quimioatrayente, y C5b, parte del complejo de ataque a la membrana que lisa las células diana. En pacientes con VAA, el depósito del complemento es evidente en los sitios de inflamación tisular, como los riñones, y los niveles plasmáticos elevados de complementos se correlacionan con la gravedad de la enfermedad. El avacopan contiene una molécula pequeña que se une a C5a lo que evita que se una a su receptor. Los resultados de los ensayos clínicos de la inhibición del receptor C5a con avacopan mostraron resultados prometedores. Sesenta y siete pacientes con VAA se aleatorizaron para recibir glucocorticoides en dosis altas, avacopan más glucocorticoides en dosis bajas, o avacopan solo junto con la inducción de ciclofosfamida o rituximab. A las 12 semanas, se produjo una reducción de 50% con respecto al valor inicial en el puntaje de actividad de vasculitis de Birmingham (BVAS) en 86% de los grupos de avacopan/glucocorticoide y 81% de los grupos de avacopan solo, en comparación con 70% en el grupo de glucocorticoides (p = 0.002 y p = 0.01, de forma respectiva). Sin embargo, este estudio incluyó sólo enfermedades no graves. Los resultados de un ensayo fase III, el ensayo Avacopan en pacientes con vasculitis asociada a ANCA (ADVOCATE), se reportaron en 2021. Este estudio reclutó a 331 pacientes, asignados al azar para recibir avacopan o glucocorticoides durante la inducción de la remisión con ciclofosfamida o rituximab. A las 26 semanas, el número de pacientes en remisión, evaluado por una puntuación de 0 en el BVAS y la retirada de la terapia con esteroides, no fue inferior en los grupos de avacopan y prednisona (72.3%, 70.1% de forma respectiva). Los datos adicionales mostraron que el avacopan fue superior a los glucocorticoides en remisión sostenida a las 52 semanas, con un perfil aceptable de seguridad. Un Ab monoclonal anti-C5a, IFX-1, también se evalúa en estudios fase II (ensayo INFLARX, NCT03895801 y NCT03712345). El reclutamiento está en curso y se estima que la finalización está prevista para julio de 2021.
3.7. Otros objetivos
Interferón-α
Los interferones (IFN), una gran familia de glicoproteínas, producen una respuesta celular a los microbios, tumores y antígenos. Se demostró que el IFN-α modula el equilibrio Th1/Th2 hacia Th1 lo que aumenta la producción de IFN-γ e inhibe la producción de IL-5 e IL-13 en las células Th2.
La eficacia del IFN-α está bien establecida en la EB, con datos provenientes de series de casos, en especial en manifestaciones oculares. Un estudio retrospectivo reportó que no hubo diferencias entre el tratamiento con azatioprina más colchicina e IFN-α2a en la uveítis por EB con respecto a las tasas de remisión y recaída. Algunos reportes de casos también reportaron la eficacia de INF-α en neuro EB. En un ECA de 44 pacientes con EB, el tratamiento con IFN-α mejoró de forma significativa las manifestaciones mucocutáneas, como úlceras orogenitales y lesiones papulopustulosas. En un estudio prospectivo reciente de 33 pacientes con trombosis venosa profunda, una de las complicaciones graves de la EB, la tasa de recaída fue menor y la tasa de recanalización fue mayor en los pacientes tratados con IFN-α en comparación con AZA (12% contra 45% y 86% contra 45%).
4. Conclusiones
A medida que avanza la comprensión de la patogenia de la vasculitis sistémica, se proponen nuevas moléculas diana y enfoques terapéuticos. Los resultados del tratamiento de la vasculitis mejoran con varios tratamientos nuevos basados en evidencia. A pesar del éxito del bloqueo de la IL-6 en la vasculitis de vasos grandes, las tasas de recaída son altas, lo que sugiere que se necesitan más estudios. En la VAA, los ensayos recientes de terapias que se dirigen a la activación de las células B, los complementos y la IL-5 proporcionan pruebas alentadoras de mejores resultados para estos pacientes. Los ensayos clínicos futuros de estos nuevos agentes terapéuticos deberán establecer su eficacia y, a medida que se disponga de un número cada vez mayor de tratamientos potenciales, deberán indicar cómo se pueden utilizar para complementar o reemplazar los enfoques existentes.
Vasculitis: From Target Molecules to Novel Therapeutic Approaches


Centro Regional de Alergia e Inmunología Clínica CRAIC, Hospital Universitario “Dr. José Eleuterio González” UANL, Monterrey, México
Dra. Med. Sandra Nora González Díaz Jefe y Profesor
Dra. Marisela Hernández Robles Profesor
Dr. Jesús Eduardo Uc Rosado Residente 1er Año
Dra. Alejandra Macías Weinmann Profesor
No hay comentarios:
Publicar un comentario
Nota: solo los miembros de este blog pueden publicar comentarios.